Solid-phase extraction columns in the analysis of lipids
The Author: William W. Christie, James Hutton Institute (and Mylnefield Lipid Analysis), Invergowrie, Dundee (DD2 5DA), Scotland..
The following was first published by W.W. Christie, in Advances in Lipid Methodology - One, pp. 1-17 (1992) (ed. W.W. Christie, Oily Press, Ayr), and is now reproduced here by kind permission of P.J. Barnes & Associates (The Oily Press), who retain the copyright to the original article. While this review is now out-of-date in terms of applications to lipids, it remains a useful guide to the principles of solid-phase extraction and to the early literature on the subject. An up-to-date list of references can be accessed here.
- Introduction
- Extraction of Lipids from Biological Fluids
1. Gangliosides
2. Prostaglandins and other eicosanoids
3. Purification of total lipid extracts
4. Some miscellaneous extraction procedures - Adsorption Chromatography for the Isolation of Lipid Classes
1. Lipid class or group separations on silica gel
2. Adsorption chromatography with chemically bonded phases - Reversed-Phase Applications
- Silver Ion Chromatography
- References
Introduction
The term "solid-phase" or "sorbent extraction", frequently abbreviated to "SPE", simply implies a physical extraction process involving a liquid and a solid phase. In practice, it has come to mean the use of commercial pre-packed columns containing stationary phases related to those used widely in high-performance liquid chromatography (HPLC), that may be adsorbents such as silica gel or Florisil™, reversed-phase materials (e.g. with chemically bonded octadecylsilyl ("ODS" or "C18") groups) or ion-exchange media (e.g. with bonded aminopropyl or phenylsulfonic acid moieties). The packing material is held in a place within a plastic (usually polypropylene of a serological grade) column by porous frits, also constructed of a plastic material, and the column ends in a Luer tip to facilitate connection to a vacuum manifold, to a needle or to a collection vessel (Fig. 1). The columns are available from a number of reputable suppliers under various trade designations, such as Bond Elut™ (Analytichem International, Harbor City, California, U.S.A.), Sep-Pak™ (Waters Associates, Milford, MA, U.S.A.), Supelclean™ (Supelco Inc., Bellefonte, PA, U.S.A.) and many others. Most of the manufacturers will supply copious literature on their products describing the principles of the technique and innumerable applications, although few deal specifically with lipid separations in any detail. When they do, the account may be an uncritical one. One particularly good example of a general handbook is available from Analytichem International [72].
Figure 1. A solid-phase extraction column.

The principle of the separation will depend largely on the nature of the stationary phase, but will involve the conventional mechanisms of adsorption, partition or ion-exchange chromatography which have been discussed in relation to lipids elsewhere [11]. Similarly, the properties of chemically-bonded stationary phases useful in the analysis of lipids were described in the earlier work [11]. Like the stationary phases used in HPLC, those developed for SPE chromatography are prepared by a variety of different chemical procedures by individual manufacturers. In general, they are stable within a range of pH of about 2 to 7.5, above which the silica base is liable to dissolution; below a pH of 2.0, the silyl bond can be hydrolysed. This is rarely a problem in practice with SPE sorbents as elution times tend to be short and the columns are intended for single use. One important difference between HPLC stationary phases and those used in SPE columns is that the latter are usually made from larger silica particles, commonly about 40 µm in diameter with pore sizes of 60 Angstroms and of irregular shape to permit a rapid flow of solvent through the sorbent bed.
The objective of the analyst is ideally to isolate a component of interest from a more complex sample in a pure concentrated state. This might be achieved by choosing conditions so that the required analyte is retained on the column while the impurities pass straight through, or conversely by allowing the analyte to elute through while the impurities are retained. In some applications of SPE columns in lipid analysis, this ideal objective can indeed be attained. On the other hand, many lipids are rather similar in their physical properties and it may only be practicable to isolate groups of lipid classes of related polarity. With any new technique of proven convenience, there is often a temptation to push the separating powers or capacity of the medium up to and occasionally beyond its reproducible limits. Lipid analysts have on occasion been guilty of this with SPE technology, as is discussed below. In adsorption chromatography especially, an SPE column packed with only 0.5 gram or so of adsorbent cannot be expected to exhibit a high degree of resolution in terms of numbers of theoretical plates. Ultimately, the most useful methods will prove to be those in which the selectivity of specific mobile and stationary phases are utilized to the full.
When an aqueous mobile phase is required, one important practical difference between the use of SPE and HPLC columns packed with apparently-related stationary phases is not always recognised by newcomers to the former, i.e. a necessity to condition the columns by solvation before use if reproducible results are to be obtained. This is especially important with the more hydrophobic stationary phases such as those of the ODS type, but it has been recommended that the only sorbent not to require solvation in this way is unbonded silica [72].
At the molecular level, a bonded material consists of a foundation of siloxane groups to which the organic moieties are attached. The organic groups have the primary function in effecting most separations, but the presence of the siloxane bridges affords a potential for secondary interactions that may be relevant in some circumstances. When the first mobile phase is to be a polar one, solvation of the sorbent with a polar solvent, such as methanol, by passing several bed volumes through the column in effect wets the silica surface. Once the surface has been solvated, the surplus methanol is removed by passing the mobile phase to be used for the separation through the column. It is believed that an important physical effect of solvation is to organize the organic moieties of the stationary phase in a regular array that is more accessible to solutes in the mobile phase, as illustrated schematically in Figure 2. Acetonitrile, isopropanol, tetrahydrofuran and other polar solvents can be used instead of methanol, the main requirement being compatibility with both the polar silica surface and the hydrophobic moiety; it should also be miscible with the initial mobile phase required for the analysis. When non-polar solvents are to be used as the first mobile phase, the cartridge should be conditioned by passing several bed volumes of this through it before the sample is applied. If the sorbent bed is allowed to dry out, it must be re-solvated prior to use.
Figure 2. Schematic representation of the effect of solvation on a chemically-bonded stationary phase.
With certain stationary and mobile phases, the polar silanol groups at the silica surface on solvation can interact via hydrogen bonding with analytes containing ionic and hydroxyl groups, and this may have a profound effect on separations. Similarly, under aqueous conditions, residual unbonded silanol groups are acidic in character and can interact via ionic bonds with appropriate ionic moieties, such as protonated amines, on analytes. This also can be an important factor influencing certain separations.
Among the practical points to be considered with each application is whether the flow-rate of the mobile phase should be enhanced by applying pressure from above or suction from below, for example by using one of the commercial manifolds developed for the purpose. There is no definitive answer, but without such assistance, aqueous media will tend to pass through SPE columns very slowly indeed. Enhanced flow can often be recommended with extraction procedures that are relatively simple; gravity flow is essential when the full resolving power of the sorbent is required. The materials used for the construction of SPE cartridges inevitable contain plasticisers, such as phthalates, which are strongly UV-absorbing and have chromatographic properties that can lead to interference with further analyses. For example, they can appear as spurious peaks during the analysis of fatty acid derivatives by gas chromatography [13]. Some of this material appears to be removed by washing the cartridges with non-polar solvents prior to commencing the analysis, or during the solvation of the sorbent, but the analyst should be aware of the potential difficulty. At least two manufacturers are offering SPE cartridges made of glass to eliminate the problem. It must not be assumed that sorbents that are nominally the same from different suppliers can be used interchangeably in given applications. The chemistry used in the manufacture may differ, and in particular, the ratio of organic material to free silanol groups on the surface of the support may vary. When changing brands, it is usually necessary to do trial experiments with model compounds before proceeding to real samples.
One question the analyst will inevitably ask is why use commercial SPE columns at all? It can be much cheaper to use disposable Pasteur pipettes or syringes as columns and pack them with one's own stationary phases. There are certainly many times when the latter approach will be efficient and cost effective. On the other hand, commercial SPE columns from most reputable manufacturers are packed by machines to a uniform density, a factor that can be important in achieving reproducible separations. Adsorbents such as silica gel and Florisil™, purchased in bulk and sitting on the laboratory shelf, will slowly lose activity as they pick up moisture from the atmosphere; similar SPE columns are packed in foil containers and will retain their activity indefinitely. Some of the bonded stationary phases are costly and it may be economical in the long run simply to purchase as many SPE columns of a given type as are necessary. The commercial SPE columns are highly convenient in use and are not expensive if the analyst's time is entered into the cost equation. Lastly, although the manufacturers would hardly be expected to publicise it, some SPE columns and especially those of the ODS type may be re-used several times in certain applications before being discarded.
In the following sections, those specific applications of SPE columns published to date in lipid analytical methodology are described. Many of the stationary phases that are available commercially would appear to have properties that might fit them for related purposes, but their utility has still to be explored. There is plentiful scope for the venturesome.
Extraction of Lipids from Biological Fluids
The aliphatic moieties of lipids can interact with non-polar stationary phases, such as those of the ODS type, by van der Waals or dispersive forces. Lipids in aqueous and hydrophilic media that are applied to such columns can therefore be retained, while the ionic species and other polar non-lipid impurities pass through. Although the mechanism is relatively non-selective, it can be effective nonetheless if the elution conditions are chosen carefully. The lipid material is eventually released from the sorbent bed by passing a non-polar solvent such as hexane or chloroform through it.
1. Gangliosides
One of the first and possibly one of the more enduring applications of SPE methodology was described in a paper by Williams and McCluer [76] in which Sep-Pak™ ODS columns were utilized for the isolation of gangliosides from tissue extracts. Lipids are usually extracted from tissue matrices with chloroform-methanol (2:1 by volume) by a procedure described originally by Folch et al. [24], in which the crude extract is washed with one-fourth its volume of saline solution; the common range of lipids remain in the lower chloroform layer, while non-lipid impurities are washed out with the upper aqueous layer. Unfortunately, the more complex glycolipids and especially the gangliosides are so soluble in water that they may also be lost with the aqueous layer. They can in fact be recovered by dialyzing the aqueous phase to remove the inorganic impurities and then lyophilizing, or by various other means all of which are time-consuming and liable to error. The improved SPE method [24] involved simply passing the upper aqueous phase from a Folch extract through an ODS column, which retains the gangliosides for subsequent recovery by elution with chloroform-methanol.
It is worth describing the complete procedure in some detail as it has been adapted by others to many other groups of compounds. A Sep-Pak™ C18 cartridge was cleaned and solvated by washing three times with methanol (10 mL portions) and chloroform-methanol (2:1, 20 mL portions), methanol (10 mL) again and finally with 'Folch upper phase', i.e. chloroform-methanol-water (3:48:47 by volume; 10 mL) containing 0.1M potassium chloride. The upper phase from a Folch extraction of a tissue, also containing 0.1M KCl, was washed twice through the cartridge by applying slight pressure, the salts were removed by elution with water (10 mL) and finally the gangliosides were recovered by elution with methanol (15 mL) or a similar volume of chloroform-methanol (2:1, v/v). The cartridges were washed with methanol, equilibrated again with 'Folch upper phase' and could then be re-used up to ten times. This procedure with occasional minor variations appears now to be the standard method in most laboratories for the isolation of gangliosides [78]. It was subsequently adapted for the purification of ganglioside fractions separated by DEAE-Sephadex™ column chromatography [39,43] and for the removal of silica gel from similar fractions isolated by thin-layer chromatography (TLC) [38].
2. Prostaglandins and other eicosanoids
SPE columns packed with ODS phases have proved themselves to be of immense value in the concentration and extraction of the very low levels of prostaglandins, leukotrienes and related eicosanoids generally to be found in biological fluids, since Powell first described the technique in 1980 [53,54]. The basic method involves passing acidified serum, urine, an aqueous incubation medium or a related fluid through an ODS cartridge, which retains the required compounds and from which they can be recovered by elution with methyl formate or other organic solvents. Hydrophobic interactions are the major mechanisms involved in the initial retention and separation from inorganic and other polar contaminants, but polar interactions with residual silanol groups permit the specific elution of certain potential analytes with appropriate mobile phases. With occasional modifications, such methods appear to have become the standard for the purpose. Previously, tedious solvent extraction procedures followed by ion-exchange and other forms of chromatography were used; large volumes of solvent and other reagents were required, so increasing the opportunities for contamination with foreign impurities. Procedures of this kind have been reviewed by others [5,6].
In the procedure described originally by Powell [53,54], Sep-Pak™ ODS cartridges were solvated by passing ethanol (20 mL) then water (20 mL) through them. Ethanol was added to an aqueous tissue homogenate to a concentration of 15%, this was centrifuged then acidified to a pH of about 3 (body fluids were simply acidified first), and the supernatant was passed through the ODS cartridge. The sorbent was washed with aqueous ethanol to remove polar lipid impurities, the concentration of ethanol being adjusted to optimize the eventual recovery of specific eicosanoids of interest. Methyl formate (10 mL) was then used to recover the prostaglandins. If necessary, hexane-chloroform (65:35, v/v) was utilized to recover normal and monohydroxy fatty acids before the column was washed and re-solvated for further use. This method with occasional minor modifications has been used in several laboratories [21,25,56, 57,60,63].
Some changes to the method described above appear to be necessary for the quantitative recovery of leukotrienes and lipoxins. For example, a similar extraction method was employed in conjunction with a separate adsorption chromatography step to isolate these compounds [28,35,48], while other workers preferred to use SPE columns of silica on their own [46]. A more popular approach has been to modify the extraction procedure, by buffering the aqueous extract and/or altering the conditions required for the elution of leukotrienes from the ODS cartridges [19,33,44,61,64,65,73]. For example, Bedetti and Cantafora [4,5] extracted cells with ethanol and acidified a solution of this extract, re-dissolved in methanol-water (4:1, v/v), with acetic acid before applying the sample to an ODS column. After a preliminary wash with 50% methanol, prostaglandins were eluted with 75% methanol and leukotrienes with 100% methanol. Yet others [47] applied the 'Folch upper phase' (see Section B.1 above) from an extract of brain tissue to a solvated ODS cartridge, which was subsequently eluted with water, methanol-water (1:4, v/v) and methanol-water (4:1, v/v); the last fraction contained the leukotrienes.
2,3-Dinor-thromboxane B2 and thromboxane B2, after conversion to the oxime derivatives, were isolated from urine by the novel expedient of passing through an SPE cartridge containing bonded phenylboronic acid in the trigonal form, obtained by washing with methanol and 0.1M hydrochloric acid [9,42,75]. A stable complex was formed between the borate moiety and the two hydroxyls of the thromboxane derivatives, but the other lipids including prostaglandins were washed through column. The required compounds were then recovered by elution with methanol containing 0.1 M sodium hydroxide.
3. Purification of total lipid extracts
Although it may prove to have limited applicability in the long term, an SPE method with ODS columns for the removal of non-lipid contaminants from crude extracts of polar lipids, such as those of brain lipids, appears to have some merit [40]. In essence, a crude chloroform-methanol extract was diluted with methanol-water and poured into the reservoir of an ODS column; the eluent was diluted with methanol-water and passed through the column again, then the process was repeated. The pure lipids were obtained by elution with chloroform-methanol (1:2, v/v), or the acidic complex lipids could be eluted before the remaining phospholipids, cerebrosides and cholesterol by a two-stage procedure. In addition, gangliosides could be recovered as a distinct fraction if necessary. While the procedure would appear to be of doubtful value for tissue extracts rich in simple lipids, such as triacylglycerols, it would be worth evaluating it for microbial lipids or for extracts of photosynthetic tissue, for example
4. Some miscellaneous extraction procedures
ODS cartridges have been used for the enrichment of trace levels of short- to long-chain free fatty acids in seawater [52]. The procedure involved pumping the water (up to 1 litre) through a solvated column at a rate of 5 to 30 mL/min followed by 0.01M hydrochloric acid (1 mL). The adsorbed acids were recovered by elution with 0.1 M ammonium hydroxide in water-methanol (1:1, v/v; 2 mL) followed by methanol (1 mL), and they were converted to the pentafluorobenzyl esters for analysis by gas chromatography. With the longer-chain fatty acids especially, the recoveries were found to be essentially complete. Free fatty acids in serum and fatty acids released by enzymic hydrolysis have been isolated quantitatively after passing the serum diluted with 10% acetic acid through an ODS cartridge, followed by water; the acids were recovered from the sorbent by elution with diethyl ether for derivatization and analysis by HPLC [45].
ODS cartridges have been utilized to separate isotopically labelled complex lipids from the radioactive water-soluble precursors in an aqueous incubation medium [23]. After solvation and washing of the column with chloroform-methanol (2:1, v/v), water, chloroform-methanol again and finally 'Folch upper phase' (see Section B.1 above), the reaction medium diluted with 'Folch upper phase' was passed through the column. The column was washed with further 'Folch upper phase' and water before the required pure phospholipids or glycolipids were recovered by elution with chloroform-methanol. On the other hand, platelet-activating factor (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) was not extracted quantitatively from plasma by ODS cartridges [59] (see also Section C.1 below).
Long-chain coenzyme-A thioesters are important intermediates in lipid biosynthesis and are not easily purified by conventional chromatography procedures because they have both hydrophilic and hydrophobic regions within the molecules. It has recently been shown that isotopically labelled coenzyme A esters, prepared by chemical synthesis, can be purified by an extraction procedure with SPE columns of the ODS type [68]. The ODS cartridge was first solvated by washing with methanol and then with an aqueous buffer of 3-N-morpholinopropanesulfonic acid (pH 7.4), as this was used in the medium required for the synthesis of the coenzyme A esters. The reaction mixture was passed through the column, followed by fresh buffer then methanol-water (1:1, v/v). The required coenzyme A esters were eventually recovered from the column by elution with methanol.
An analogous method was utilized for the separation of retinol and retinyl palmitate from incubation mixtures containing these compounds [51]. The aqueous incubation mixture was first passed through an ODS SPE column; methanol-water (97:3, v/v) removed the retinol and then hexane eluted retinyl palmitate. The column was re-used several times after a washing procedure. Following a hydrolysis step, unesterified cholesterol was isolated from milk powders in a similar way [69].
A published procedure for isolation of triacylglycerols from human serum is relevant but poses a problem [10]. The serum was applied directly to an Extralut 1™ column and triacylglycerols were apparently eluted with diethyl ether-ethanol (3:1, v/v); unfortunately, the authors did not demonstrate the purity of the fraction isolated which potentially might include cholesterol esters and phospholipids.
Adsorption Chromotography for the Isolation of Lipid Classes
1. Lipid class or group separations on silica gel
Lipid extracts from natural sources tend to contain many different classes or groups of compounds. There are few if any techniques that can be used to isolate each of these in a single operation. It is usually easier from a technical standpoint to obtain single lipid classes in a pure state, after a preliminary fractionation has been carried out, into simple lipid, phospholipid or glycolipid groups. Column chromatography with silica gel as the adsorbent is the simplest method of accomplishing this. SPE cartridges are especially convenient for the purpose, since the degree of hydration and thence the adsorptivity of the silica is closely controlled. Fractions isolated by this means can later be analysed in much greater detail by HPLC or TLC.
Bitman et al. [7], for example, washed a cartridge of silica gel (size not stated but probably 0.6 g) with chloroform (20 mL) to eliminate any potential impurities before the lipid sample was applied in hexane solution. The non-polar (simple) lipids, such as cholesterol esters, triacylglycerols and cholesterol, were eluted with hexane-diethyl ether (1:1, v/v; 40 mL), and the complex lipids were recovered by elution first with methanol (20 mL) and then with chloroform-methanol-water (3:5:2 by volume: 20 mL). With the lipids of milk fat, in which the complex lipid content is only 1%, as much as 100 mg of lipid could be loaded on the column, but 10 mg of complex lipids would probably be a realistic upper limit. The author has used this procedure successfully [17].
In a similar separation procedure, but one in which SPE cartridges per se were not used, chloroform eluted the simple lipids, methyl formate eluted prostaglandins, acetone eluted glycolipids and methanol eluted phospholipids [62]. Many comparable separations have been described. Hexane-diethyl ether (in which the latter component varied from 20 to 100%) [8,49], chloroform [31,32,77] and chloroform-acetic acid (100:1, v/v) [1,26,31] have been utilized for the isolation of the simple lipid classes from silica gel SPE cartridges, while the phospholipids (or complex lipids) were recovered by elution with methanol [31,32,49], chloroform-methanol (1:1, v/v) [77], methyltert-butyl ether-acetic acid (100:0.2, v/v) [27], methanol-water (98:2, v/v) and methanol-chloroform-water (2:1:0.8 by volume) [26,31]. In one instance [77], an intermediate elution step with acetone-methanol (9:1, v/v) was used to isolate a distinct glycolipid fraction. No one has yet compared all of these critically, although Janero and Burghardt [31] have made a valuable start, and it is not easy to offer critical comment because different brands of SPE cartridge and volumes of solvent were employed, and various samples were analysed in each of the papers cited.
In addition to these group separations, the convenience of SPE cartridges has tempted many analysts to attempt more refined separations of lipid classes. For example, a simple elution scheme involving elution first with methanol-chloroform (2:1, v/v) then methanol-chloroform-water (2:1:0.8 by volume) gave fractions enriched in phosphatidylethanolamine and phosphatidylcholine, respectively [26]. This procedure was evaluated by others [1,31] and was found to be especially suitable for the extraction of the biologically-active lipid, platelet-activating factor [31]. A second procedure [27] was developed from this in which cholesterol esters and triacylglycerols were each eluted with hexane and methyltert-butyl ether in the ratios 100:1.5 and 96:4 by volume, respectively; the column was acidified by a wash with hexane-acetic acid (100:0.2, v/v) and free acids were recovered with hexane-methyltert-butyl ether-acetic acid (100:2:0.2 by volume). After free cholesterol had been recovered by elution with methyltert-butyl ether-acetic acid (100:0.2, v/v), various fractions enriched in particular phospholipids were eluted with mobile phases of increasing polarity containing methyltert-butyl ether, methanol and an ammonium acetate buffer (pH 8.6) in various proportions. The author has heard anecdotal reports that this procedure has not been easy to reproduce in other laboratories, especially for each of the simple lipids. It appears that the resolving power of the small columns is being pushed to its limit, and it is not surprising that small changes in local conditions, e.g. temperature, humidity and batch variations in the sorbent, may conspire to defeat the procedure. In addition, it has been reported that the basic conditions used for the elution of the phospholipids can lead to degradation of platelet-activating factor [31].
It has also been claimed that hexane-diethyl ether (98.5:1.5, v/v) will elute a clean cholesterol ester fraction of serum lipids from an SPE cartridge of silica gel [74]. Among other miscellaneous separations, hexane-diethyl ether (95:5, v/v) was used in the purification of triacylglycerols [49,50] and chloroform-methanol (98:2, v/v) eluted methylated prostaglandins from a chromatographic fraction enriched in these compounds [48]. Similar methodology has been utilized to isolate leukotrienes [28,46]. In a novel procedure for the isolation of the free fatty acids in brain lipid extracts, they were converted to the ammonium salts; chloroform saturated with ammonium hydroxide eluted the bulk of the simple lipids, then chloroform-acetic acid eluted the unesterified fatty acid fraction [1]. On the other hand, most analysts appear to prefer SPE columns with aminopropyl groups chemically bonded to silica for this purpose (see below).
2. Adsorption chromatography with chemically bonded phases
Commercial SPE columns are available with a wide range of chemically bonded stationary phases. These must have great potential for the isolation of specific lipid classes, yet surprisingly few applications have been described to date. Kaluzny et al. [34] were among the first to apply SPE cartridges packed with aminopropyl bonded phases for separating groups of lipid classes, including non-acidic phospholipids. Unfortunately, their claims of quantitative recovery of phospholipids were in error. For example, acidic phospholipids, such as phosphatidylinositol and phosphatidic acid, are retained very strongly [17,22,36], and as is true for similar packings in HPLC and other column chromatography applications (reviewed elsewhere [11]), solvents of high ionic strength are required to elute them. For example, a modified method appears to be of value for the isolation of phosphatidylglycerol from lung surfactant [22]; methanol was used to elute the non-acidic phospholipids, then a mobile phase containing a high concentration of ionic species, i.e. dichloromethane-methanol-ammonium hydroxide (28%)-ammonium acetate in methanol (10 mmol/L) in the proportions 28:7:1:1 by volume, brought off the required phosphatidylglycerol. Finally, phosphatidylserine and phosphatidylinositol were recovered by elution with hexane-isopropanol-water-ammonium hydroxide (60:60:20:1 by volume). Others [36] have devised an alternative procedure using such SPE columns for the quantitative isolation of simple lipid, free fatty acid, neutral phospholipid (plus cerebroside) and acidic phospholipid fractions. It would also be of value to determine whether a clean glycolipid fraction could be obtained by eluting with a solvent such as acetone prior to recovering the neutral phospholipids.
Isolation of individual simple lipid classes by stepwise elution from such columns was also described [34], but again this has not been easy to reproduce in other laboratories (apart from an unesterified fatty acid fraction), as with a similar procedure described by others [29]. A further modification of the procedure of Kaluzny et al. made a virtue of what might be considered a disadvantage, i.e. that acidic lipids are strongly retained, since it enabled the elution of fractions enriched in cholesterol and phosphatidylcholine for determination by specific enzymatic procedures [2].
Aminopropyl SPE cartridges are especially useful for the isolation of the free fatty acid fraction from lipid extracts [20,22,34,55]. Chloroform-isopropanol (2:1, v/v) was used first to elute most of the simple lipids, before the free fatty acids were recovered quantitatively with diethyl ether-acetic (or formic) acid (98:2, v/v).
Eicosanoids may also elute in the latter solvent [6], and this property has been utilized in a method for the isolation and estimation of leukotriene E4 [35].
What further opportunities exist for SPE columns packed with bonded phases? Various alternative amine phases are available that may be of value for the isolation of prostaglandins, phospholipids and glycolipids. SPE columns in which the important functional group is a nitrile, carboxyl or sulfonic acid can be purchased; structurally related phases are widely used in HPLC and other column chromatography procedures for the separation of complex lipids and might be of special value for acidic lipids. For example, home-made mini-columns packed with Amberlite™ XAD-2 ion-exchange resin have been employed for the quantitative extraction of platelet-activating factor from plasma [59]. Phenylboronic acid cartridges (Bond Elut™) have been utilized for the isolation of oligosaccharides [66], but apparently not yet for glycolipids or partial glycerides, for example. The potential is evident from a related non-commercial system that has been described [37].
D. Reversed-Phase Applications
Although the resolution expected in terms of numbers of theoretical plates from SPE columns packed with ODS phases is not great, it can be sufficient to separate lipids and other hydrophobic compounds that are very different in terms of molecular weight. Again the potential of the technique is probably greater than is apparently indicated by the few published applications. For example, Bond Elut™ ODS cartridges were utilized as part of a benzoylation procedure for gangliosides to remove the excess derivatizing reagents [71]. After evaporation of the solvents used for the reaction, the products were washed through the cartridge with methanol, which eluted the excess benzoylating reagent (predominantly benzoyl chloride), then the benzoylated gangliosides were recovered with methanol-benzene (8:2, v/v). Similar methods were used in the purification of benzoyl derivatives of eicosanoids [3], and of dinitrobenzoyl [67] and diastereomeric naphthylethyl urethane derivatives of diacylglycerols [16]. With the last, the reaction mixture was applied to a solvated ODS cartridge in methanol-water (95:5, v/v) to eliminate the unwanted by-products before the required compounds were eluted with acetone. After re-equilibration with the starting solvent, the cartridges could be re-used repeatedly. A further application, that might have been discussed equally under Section B above, was for the elimination of urea from the non-urea adducted fraction (cyclic fatty acid monomers) of hydrogenated methyl ester derivatives prepared from edible fats and oils [58].
That the technique has some potential for more subtle separations is confirmed by a report of the enrichment of methyl eicosapentaenoate from methyl esters of algal fatty acids [18]. This compound eluted ahead of C16 and C18 fatty acids (1 mg scale) from an ODS cartridge with acetonitrile-water at various concentrations as the mobile phase.
E. Silver Ion Chromotography
The technique of silver ion or 'argentation' chromatography has proved of enormous value to lipid analysts for the separation of molecular fractions differing in degree of unsaturation. In much of the published work, TLC has been used, but this has a number of disadvantages; in particular, the technique is messy and silver salts are co-eluted with fractions of interest in preparative applications. HPLC is now being used more often in the silver ion mode, especially in conjunction, with ion-exchange phases [12], but a substantial capital investment in equipment is needed.
SPE columns of silica gel impregnated with silver nitrate have been utilized for the separation of fatty acid esters of diterpenoids [41], but contamination of fractions with silver ions is likely and better methods are now available. The author [14] showed that Bond Elut™ columns packed with a silica-based benzenesulfonic acid medium could be converted to the silver ion form and used to achieve excellent separations of methyl ester derivatives of fatty acids. No silver salts were eluted concomitantly. A solution of silver nitrate (20 mg) in acetonitrile-water (0.25 mL; 10:1, v/v) was allowed to flow through a Bond Elut™ SCX cartridge, wrapped to the level of the top of the sorbent bed in aluminium foil to exclude light; the SPE column was flushed with acetonitrile (5 mL), acetone (5 mL) and dichloromethane (10 mL) and was then ready for use. These solvents in various proportions were also employed in the optimum elution scheme for the isolation of fractions as listed in Table 1. A methyl ester sample (0.1 to 0.5 mg) was applied to the column in a small volume of dichloromethane. Solvent mixtures were allowed to flow under gravity, and satisfactory resolution of components with zero to six double bonds was obtained. Substantial changes in solvent composition were utilized at each step, and there was very little cross-contamination especially with the early fractions.
| Table 1. Solvent elution scheme for the recovery of the methyl ester derivatives of fatty acids (0.5 mg) of bovine testis from a Bond Elute™ SCX solid-phase extraction column in the silver ion mode [14] | |||||
| No | Solvents(%)a | Volume | Fraction | ||
| A | B | C | |||
| 1 | 100 | 5mL | saturated | ||
| 2 | 90 | 10 | 5mL | monoenes | |
| 3 | 100 | 5mL | dienes | ||
| 4 | 97 | 3 | 10mL | trienes | |
| 5 | 94 | 6 | 10mL | tetraenes | |
| 6 | 88 | 12 | 5mL | pentaenes | |
| 7 | 60 | 40 | 5mL | hexaenes | |
| aA, dichloromethane; B, acetone; C, acetonitrile | |||||
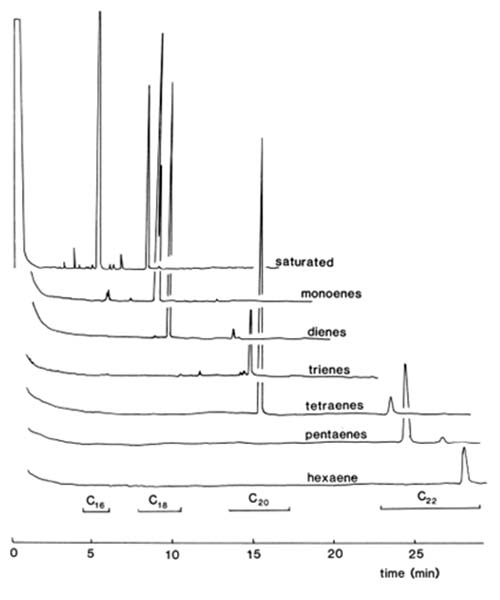
The GC traces from methyl esters derived from bovine testis lipids separated by this method are illustrated in Figure 3 to confirm the nature of the separation. It is important that the columns should not be overloaded or that the flow-rate be increased artificially otherwise resolution is lost. Further details of this procedure with a discussion of newer developments are available on this website here...
Figure 3. GC traces of fractions from a separation of methyl esters of fatty acids (0.5 mg) from bovine testis lipids on a Bond Elut™ SCX SPE column in the silver ion form and eluted under the conditions of Table 1 (reproduced by kind permission of the Journal of Lipid Research [14]).
A very similar method was described independently by others [70]. The method has been adapted for the isolation and quantification of the molecular species of triacylglycerols from relatively saturated fats [15] and for cholesterol esters [30]. With the former, dichloromethane-methyl acetate mixtures gave good resolution of species [15]. On the other hand, cholesterol esters were eluted under the same conditions as the corresponding methyl esters, since the double bond in the sterol moiety did not appear to influence the separation [30]. More applications of this kind will undoubtedly be described in the future.
ABBREVIATIONS: - SPE, solid-phase extraction; HPLC, high-performance liquid chromatography; TLC, thin-layer chromatography; ODS, octadecylsilyl.
References
- Ansari, K.A. and Shoeman, D., J. Chromatogr., 439, 453-458 (1988).
- Aufenanger, J. and Katterman, R., J. Clin. Chem. Clin Biochem., 27, 605-611 (1989).
- Baertsh, S.W., Ingram, C.D., Harris, T.M. and Brash, A.R., Biochemistry, 27, 18-24 (1988).
- Bedetti, C. and Cantafora, A., In: Production & Exploitation of Existing and New Animal Cell Substrates, pp. 331-336 (1984) (edited by E.R. Spier, B. Griffiths & W. Hennessen, Karger, Basel).
- Bedetti, C. and Cantafora, A., Adv. Biochem. Eng./Biotechnol., 35, 47-81 (1987).
- Birkle, D.L., Bazan, H.E.P. and Bazan, N.G. In: Neuromethods 7. Lipids and Related Compounds, pp. 227-244 (1988) (edited by A.A. Boulton, G.B. Baker & L.A. Horrocks, Humana Press, Clifton).
- Bitman, J., Wood, D.L., Hamosh, M., Hamosh, P. and Mehta, N.R., Am. J. Clin. Nutr., 38, 300-312 (1983).
- Blunk, H.-C. and Steinhart, H., Z. Lebensm. Unters. Forsch., 190, 123-125 (1990).
- Catella, F. and Fitzgerald, G.A., Methods Enzymol., 187, 42-50 (1990).
- Chaves das Neves, H.J., Vasconcelos, A.M.P. and Vasconcelos, O., J. High Resolut. Chromatogr., 13, 520-523 (1990).
- Christie, W.W., Lipid Analysis 3rd Edition. (Oily Press, Bridgwater) (2002).
- Christie, W.W., J. High Resolut. Chromatogr. Chromatogr. Commun., 10, 148-150 (1987).
- Christie, W.W., Gas Chromatography and Lipids (1989) (The Oily Press, Ayr).
- Christie, W.W., J. Lipid Res., 30, 1471-1473 (1989).
- Christie, W.W., J. Sci. Food Agric., 52, 573-577 (1990).
- Christie, W.W., Nikolova-Damyanova, B., Laakso, P. and Herslof, B., J. Am. Oil Chem. Soc., 68, 695-701 (1991).
- Christie, W.W., Noble, R.C. and Davies, G., J. Soc. Dairy Technol., 40, 10-12 (1987).
- Cohen, Z. and Cohen, S., J. Am. Oil Chem. Soc., 68, 16-19 (1991).
- Dawson, M. and McGee, C., J. Chromatogr., 532, 379-386 (1990).
- De Jong, C.and Badings, H.T., J. High Resolut. Chromatogr., 13, 94-98 (1990).
- Doehl, J. and Greibrokk, T., J. Chromatogr., 529, 21-32 (1990).
- Egberts, J. and Buiskool, R., Clin. Biochem., 34, 163-164 (1988).
- Figlewicz, D.A., Nolan, C.E., Singh, I.N. and Jungalwala, F.B., J. Lipid Res., 26, 140-144 (1985).
- Folch, J., Lees, M. and Stanley, G.H.S., J. Biol Chem., 226, 497-509 (1957).
- Ford-Hutchinson, A.W., Piper, P.J. and Samhoun, M.N., Br. J. Pharmacol., 76, 215-220 (1982).
- Hamilton, J.G. and Comai, K., J. Lipid Res., 25, 1142-1148 (1984).
- Hamilton, J.G. and Comai, K., Lipids, b, 1146-1149 (1988).
- Hofmann, U., Seefried, S., Meese, C.O., Mettang, T., Hubel, E. and Kuhlmann, U., Anal. Biochem., 189, 244-248 (1990).
- Hoving, E.B., Jansen, G., Volmer, M., van Doormaal, J.J. and Muskiet, F.A.J., J. Chromatogr., 434, 395-409 (1988).
- Hoving, E.B., Muskiet, F.A.J. and Christie, W.W., J. Chromatogr., 565, 103-110 (1991).
- Janero, D.R. and Burghardt, C., J. Chromatogr., 526, 11-24 (1990).
- Juaneda, P. and Rocquelin, G., Lipids, 20, 40-41 (1985).
- Jubiz, W., Nolan, G. and Kaltenborn, K.C., J. Liq. Chromatogr., 8, 1519-1526 (1985).
- Kaluzny, M.A., Duncan, L.A., Merritt, M.V. and Epps, D.E. J. Lipid Res., 26, 135-140 (1985).
- Kikawa, Y., Nakai, A., Shigematsu, Y. and Sudo, M., J. Chromatogr., 532, 387-393 (1990).
- Kim, H.-Y. and Salem, N., J. Lipid Res., 31, 2285-2289 (1990).
- Krohn, K., Eberlein, K. and Gercken, G., J. Chromatogr., 153, 550-552 (1978).
- Kubo, H. and Hoshi, M., J. Lipid Res., 26, 638-641 (1985).
- Kundu, S.K. and Suzuki, A., J. Chromatogr., 224, 249-256 (1981).
- Kyrklund, T., Lipids, 22, 274-277 (1987).
- Lam, L.K.T., Yee, C., Chung, A. and Wattenberg, L.W., J. Chromatogr., 328, 422-424 (1985).
- Lawson, J.A., Brash, A.R., Doran, J. and FitzGerald, G.A., Anal. Biochem., 150, 463-470 (1985).
- Ledeen, R.W. and Yu, R.K., Methods Enzymol., 83, 139-191 (1982).
- Luderer ,J.R., Riley, D.L. and Demers, L.M., J. Chromatogr., 273, 402-409 (1983).
- Matsuzawa, T., Mishima, K., Nishii, M. and Ito, M., Biochem. Int., 15, 693-702 (1987).
- Metz, S.A., Hall, M.E., Harper, T.W. and Murphy, R.C., J. Chromatogr., 233, 193-201 (1982).
- Miyamoto, T., Lindgren, J.A. and Samuelsson, B., Biochim. Biophys. Acta, 922, 372-378 (1988).
- Muller, H., Mrongovius, R. and Seyberth, H.W., J. Chromatogr., 226, 450-454 (1981).
- Nash, A.M. and Frankel ,E.N., J. Am. Oil Chem. Soc., 63, 244-246 (1986).
- Neff, W.E., Frankel, E.N. and Miyashita, K., Lipids, 25, 33-39 (1990).
- O'Connor, C.J. and Yaghi, B., J. Lipid Res., 29, 1693-1697 (1988).
- Pempkowiak, J., J. Chromatogr., 258, 93-102 (1983).
- Powell, W.S., Prostaglandins, 20, 947-957 (1980).
- Powell, W.S., Methods Enzymol., 86, 467-477 (1982).
- Prasad, M.R., Jones, R.M., Young, H.S., Kaplinsky, L.B. and Das, D.K., J. Chromatogr., 428, 221-228 (1988).
- Raghunath, M., Stiegeler, A., Lange, B. and Frosch ,P., J. Liq. Chromatogr., 13, 969-980 (1990).
- Ramis, I., Rosello-Catafau, J., Artigot, M., Bulbena, O., Picado,C. and Gelpi, E., J. Chromatogr., 532, 217-225 (1990).
- Rojo, J.A. and Perkins, E.G., J. Am. Oil Chem. Soc., 66, 1593-1595 (1989).
- Salari, H., J. Chromatogr., 382, 89-98 (1986).
- Salari, S.H., De Voe, I.W. and Powell, W.S., Biochem. Biophys. Res. Commun., 104, 1517-1524 (1982).
- Salari, H. and Stephenrud, S., J. Chromatogr., 378, 35-44 (1986).
- Saunders, R.D. and Horrocks, L.A., Anal. Biochem., 143, 71-75 (1984).
- Shak, S. and Goldstein, I.M., J. Biol. Chem., 259, 10181-10187 (1984).
- Schulz, R. and Seeger, W., Biochem. Pharmacol., 35, 183-193 (1986).
- Steinhilber, D., Herrman, T. and Roth, H.J., J. Chromatogr., 493, 361-366 (1989).
- Stoll, M.S. and Hounsell, E.F., Biomed. Chromatogr., 2, 249-253 (1988).
- Takamura, H., Tanaka, K., Matsuura, T. and Kito, M., J. Biochem. (Tokyo), 105, 168-172 (1989).
- Taylor ,D.C., Weber, N., Hogge, L.R. and Underhill, E.W., Anal. Biochem., 184, 311-316 (1990).
- Tsui, I.C., J. Assoc. Off. Anal. Chem., 72, 421-424 (1989).
- Ulberth, F. and Achs, E., J. Chromatogr., 504, 202-206 (1990).
- Ullman, M.D. and McCluer, R.H., J. Lipid Res., 26, 501-506 (1985).
- Van Horne, K.C. (editor) Sorbent Extraction Technology (1985) (Analytichem International, Harbor City, CA).
- Verhagen, J., Wassink, J.A., Kijne, G.M., Vietor, R.J. and Bruynzeel, P.L.B., J. Chromatogr., 378, 208-214 (1986).
- Wang, S.T. and Peter, F., J. Chromatogr., 276, 249-256 (1983).
- Weber, C., Holler, M., Beetens, J., De Clerk, F. and Tegtmier, F., J. Chromatogr., 562, 599-611 (1991).
- Williams, M.A. and McCluer, R.H., J. Neurochem., 35, 266-269 (1980).
- Yao, J.K. and Rastetter, G.M., Anal. Biochem., 150, 111-116 (1985).
- Yates, A.J., in Neuromethods 7. Lipids and Related Compounds, pp. 265-327 (1988) (edited by A.A. Boulton, G.B. Baker & L.A. Horrocks, Humana Press, Clifton).
A more recent review article is - Ruiz-Gutierrez, V. and Perez-Camino, M.C. Update on solid-phase extraction for the analysis of lipid classes and related compounds. J. Chromatogr. A, 885, 321-341 (2000); DOI: 10.1016/S0021-9673(00)00181-3.
In This Section
- Solid-phase extraction columns in the analysis of lipids
- Preparation of Ester Derivatives of Fatty Acids for Chromatographic Analysis
- Preparation of Lipid Extracts Tissues
- The Chromatographic Resolution of Chiral Lipids
- Detectors for HPLC of Lipids with Special Reference to Evaporative Lght-Scattering Detection
- Why Doesn't Your Method Work When I Try It?
- Laboratory Accreditation in a Lipid Analysis Context
- What Column do I Need for Gas Chromatographic Analysis of Fatty Acids?
- Fatty Acid Analysis by HPLC
- Alternatives to Methyl Esters for GC Analysis of Fatty Acids
- A Practical Guide to the Analysis of Conjugated Linoleic Acid (CLA)
- Application of Infrared Spectroscopy to the Rapid Determination of Total Saturated, trans, Monounsaturated, and Polyunsaturated Fatty Acids
- The Use of Lithiated Adducts for Structural Analysis of Acylglycerols by Mass Spectrometry with Electrospray Ionization
- Identification of FAME Double Bond Location by Covalent Adduct Chemical Ionization (CACI) Tandem Mass Spectrometry
- The Use of Countercurrent Chromatography (CCC) in Lipid Analysis
- Gas Chromatographic Analysis of Plant Sterols
- Analysis of Tocopherols and Tocotrienols by HPLC
- Reversed-Phase HPLC of Triacylglycerols
- Structural Analysis of Triacylglycerols
- Thin-Layer Chromatography of Lipids
- High-temperature Gas Chromatography of Triacylglycerols
- Modification of an AOCS Official Method for Crude Oil Content in Distillers Grains and Other Agricultural Materials
- Lipidomics - A Personal View