Reversed-Phase HPLC of Triacylglycerols
The Author: William W. Christie, James Hutton Institute (and Mylnefield Lipid Analysis), Invergowrie, Dundee (DD2 5DA), Scotland.
Abstract: The resolution of molecular species of triacylglycerols by reversed-phase high-performance liquid chromatography is discussed. Particular attention is given to the choice of stationary phase, mobile phase, injection solvent, detector and the effect of temperature. The influence of molecular structure on retention time is also explained.
Triacylglycerols are major components of the human diet, so methods for their analysis are of great importance. Excellent resolution of triacylglycerols by reversed-phase high-performance liquid chromatography has been possible for many years, but many practical and theoretical aspects of the process are poorly understood, although excellent reviews have been available on the topic [1,2]. With some honourable exceptions little of interest is now being published in this area. Indeed, many chromatograms continue to be illustrated that are well below the standard that should be attainable. I suspect that this is because many analysts are concerned simply with obtaining a "recipe" to do the job. A little knowledge of the theory can help enormously to improve results.
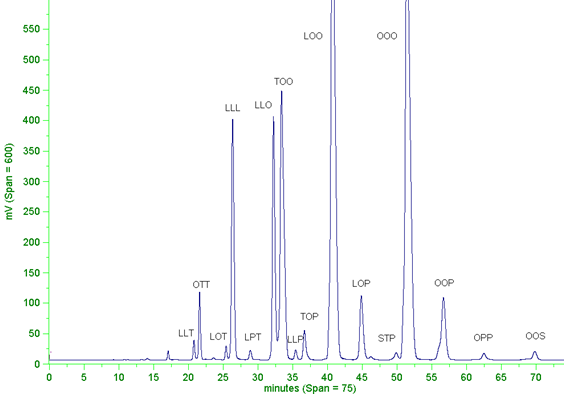
The nature of the separation is fairly well known, i.e. that the retention time is dependent on the total number of carbon atoms in the three fatty acyl moieties, with each double bond reducing the effective chain length of the species by the equivalent of about two carbon atoms. This figure "two" is sometimes interpreted too literally, and I will say more below. Figure 1 illustrates a separation of molecular species of triacylglycerols from a vegetable oil to show the nature of the separation and the quality of the resolution that should be attainable in all laboratories.
Figure 1. Separation of triacylglycerols from a low-erucic acid rapeseed oil by reversed-phase HPLC (Spherisorb ODS2; 250 × 4.6 mm column) with a gradient of acetone into acetonitrile (1 mL/min) and evaporative light-scattering detection (Abbreviations: P = palmitate, S = stearate, O = oleate, L = linoleate, T = triene (linolenate).
Stationary Phase
There is a limited choice of stationary phases that we need consider. Phases of the octadecylsilyl (C18 or ODS) type, chemically bonded to silica, are used almost universally, and those with a high-carbon loading appear to be best (often termed "ODS2"). From a theoretical standpoint, a stationary phase similar in chain length to the fatty acyl chains maximizes the interactions and should give the highest efficiency (C8 and C30 columns have been tried with no obvious advantage). There may be differences between particular commercial makes of phase, but there is insufficient information to discuss this properly. In any case, these will change with continuing developments in production methods.
The 'standard' 250 × 4.6 mm column with 5 micron packing can easily be replaced by, say, a 100 × 3 mm column with a 3 micron packing, with savings in terms of time and solvent, and with no loss of resolution. Indeed, so called ‘ultra-high-performance liquid chromatography’ procedures with 100 × 2.1 mm columns and 1.7 micron packings have given excellent results with mass spectrometric [3] and evaporative light-scattering detection [4].
Mobile Phase
Propionitrile is the only pure solvent that has been widely used as a mobile phase, but it is expensive (although depending on the chromatographic system, it may be recoverable) and highly toxic. Nowadays, it is mainly reserved for HPLC-mass spectrometry applications.
A more practical alternative for most of us is to use acetonitrile as the main component of the mobile phase, with a further solvent added to modify the elution properties and optimize the separation of the triacylglycerol analytes. It is the choice of modifier solvent that causes analysts most problems. Indeed, I know of no objective means of assessing which one to recommend, since each one has a different solvent strength and different amounts of each may have to be added to obtain elution of specific components in a given time. Certainly there have been only a few noteworthy attempts to make objective comparisons.
The modifier solvent serves two functions, i.e. it improves the solubility of triacylglycerols in the mobile phase, and it has an effect on the selectivity and efficiency of the separation. In my experience, acetone-acetonitrile mixtures are very good for the common range of vegetable oils, but are less well suited to milk fat, say, because the saturated components of higher molecular weight, such as tristearin, tend to precipitate out of solution. This effect is not seen with dichloromethane-acetonitrile mixtures, which have been used in many of the better separations that have been published, and at least one systematic study indicates that this solvent combination may be the best available for the purpose [5]. However, dichloromethane has a rather low boiling point, and on a hot summer's day with the sun streaming in the window and a lot of electrical equipment in use, it can start to evaporate rather quickly (even in Scotland). The proportion in the mobile phase will change, and sometimes a vapour lock in the HPLC pump occurs. Good results have also been obtained with tetrahydrofuran-acetonitrile mixtures, although methyl tert-butyl ether may now be preferred over tetrahydrofuran as it is more stable to oxidation.
It is my impression that acetone, dichloromethane and tetrahydrofuran added to acetonitrile in proportions that elute standards in similar times have comparable effects on the selectivity of the separation, i.e. triacylglycerol components always emerge in the same order. However, the efficiency of the separation may not be exactly the same.
Of course, the other component of the chromatographic system that may determine the choice of mobile phase is the detector. Tetrahydrofuran (or methyl tert-butyl ether) and propan-2-ol are in essence the only modifier solvents that can be used with UV detection.
Injection Solvent
One important practical point that should be mentioned is the choice of injection solvent. Samples are best injected in the minimum volume of the mobile phase, i.e. 5 to 10 microlitres, and the injection loop in the Rheodyne valve for manual injection should be of this size. If the sample is insufficiently soluble in this volume of the mobile phase, then the modifier component of the phase should be used.
In no circumstances should hexane be employed as the injection solvent. It is so similar in its properties to the stationary phase that it competes with this for the solute molecules, causing peak broadening and it can even cause single components to emerge as double peaks. Unfortunately, hexane is used in so many other chromatographic systems as the injection solvent that novices to HPLC tend to use it in reversed-phase analyses without thinking.
Column Temperature
Column temperature is very important, and I am convinced of the value of temperature control whenever this is feasible. Most analysts use ambient temperature, which is of course a rather variable commodity. I found this to my cost once in running a large number of samples with autoinjection overnight; retention times almost doubled when the heating went off in the laboratory in the evening. If the column temperature can be fixed by installing it in a thermostatted oven, much more reproducible retention times are achieved. In theory, the lower the temperature, the better the separation. However, solubility effects may come into play and if some of the more saturated molecular species precipitate out of solution, the separation will worsen. The optimum column temperature for any given sample has to be determined by trial and error.
Detector
I am reluctant to recommend any specific detection system for quantitative analysis of triacylglycerol species separated by HPLC. Refractive index detection can give good results (if you have a high-quality instrument), and a collaborative study has confirmed this [6]. UV detection at 220 nm has also been reported to give good quantitative data, with acetonitrile-tetrahydrofuran as mobile phase, but a major collaborative study is required for confirmation. Both of these detectors are best with isocratic elution. UV detection at lower wavelengths is of little value for quantification purposes. The evaporative light-scattering detector gives excellent semiquantitative results, but it must be calibrated with great care for higher precision. In particular, the response tends to decrease as peaks widen, as is inevitable from the nature of the detection system. Any calibration method must take this into account.
Mass spectrometry is of course an invaluable means both for detecting and identifying molecular species of triacylglycerols. Atmospheric-pressure chemical ionization (APCI) is well suited to identification of components, but is not ideal in terms of quantification unless great care is taken in calibration [3]. Mass spectrometry with electrospray ionization may be best overall in terms of sensitivity and quantification, although there is still a need for careful calibration [7,8].
Of course, in research applications with limited numbers of samples, it is always possible to collect fractions, add an internal standard, and transesterify for analysis by gas chromatography for simultaneous identification and quantification [8].
Separation Principle
As I discussed briefly earlier, a double bond in the fatty acyl chain reduces the retention time by the equivalent of about two carbon atoms. In fact in efficient modern systems, the exact figure is less than two and depends on the position of the double bond. The double bonds in a diene have a different effect from those in two monoenes. The old concept of partition number (PN) in which the number of carbon atoms in the fatty acyl chains was summed, then 2 was subtracted for each double bond, has very little meaning therefore. To give just one example, the triacylglycerol species, 18:1-18:1-18:2 (PN = 46) elutes before 14:0-14:0-16:0 (PN = 44).
A number of useful theoretical treatments of elution order have been published that utilize thermodynamic considerations to provide convincing explanations. However, I found that my most useful and practical source of information was a comprehensive table of elution values, published by Perrin and Naudet [9]. Once the main components in an unknown vegetable oil are identified by comparison with more familiar oils, it is a relatively easy matter to consult the table and decide which of the various possibilities are plausible from the known fatty acid composition.
There are times when I prefer silver ion HPLC to the reversed-phase technique and vice versa, or when the two techniques should be used sequentially. If we are to make the best use of the latter technique, attention to detail will certainly help.
References
- Nikolova-Damyanova, B. Reversed-phase high-performance liquid chromatography: general principles and applications to the analysis of fatty acids and triacylglycerols. In Advances in Lipid Methodology - Four, pp. 193-251 (ed. W.W. Christie, Oily Press, Dundee) (1997).
- Heron, S., Bleton, J. and Tchapla, A. Mechanism for separation of triacylglycerols in oils by liquid chromatography: identification by mass spectrometry. In New Trends in Lipid and Lipoprotein Analyses, pp. 205-231 (ed. J.-L. Sébédio and E.G. Perkins, AOCS Press, Champaign, USA) (1995).
- Leskinen, H.M., Suomela, J.P. and Kallio, H.P. Quantification of triacylglycerol regioisomers by ultra-high-performance liquid chromatography and ammonia negative ion atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom., 24, 1-5 (2010) (DOI: 10.1002/rcm.4346).
- Ross, K.L., Hansen, S.L. and Tu, T. Reversed-phase analysis of triacylglycerols by ultra performance liquid chromatography-evaporative light scattering detection (UPLC-ELSD). Lipid Technol., 12, 14-16 (2011) (DOI: 10.1002/lite.201100083).
- Heron, S. and Tchapla, A. Choice of stationary and mobile phases for separation of mixed triglycerides by liquid-phase chromatography. Analusis, 22, 114-126 (1994).
- Firestone, D.J. Liquid-chromatographic method for determination of triglycerides in vegetable oils in terms of their partition numbers - summary of collaborative study. Ass. Off. Anal. Chem. Int., 77, 954-957 (1994).
- Byrdwell, W.C. Qualitative and quantitative analysis of triacylglycerols by atmospheric pressure ionization (APCI and ESI) mass spectrometry techniques. In Modern Methods for Lipid Analysis by Liquid Chromatography/Mass Spectrometry and Related Techniques. pp. 298-412 (ed. W.C. Byrdwell, AOCS Press, Champaign) (2005).
- Christie, W.W. and Han, X. Lipid Analysis - Isolation, Separation, Identification and Lipidomic Analysis (4th edition), 446 pages (Oily Press, Bridgwater, U.K.) (2010) - www.pjbarnes.co.uk/op/la4.htm.
- Perrin, J.-L. and Naudet, M. Identification et dosage des triglycérides des corps gras naturels par CLHP. Rev. Franç. Corps Gras, 30, 279-285 (1983).
This article has been updated appreciably from one first published by the author in Lipid Technology, 8, 140-142 (1996) (by kind permission of P.J. Barnes & Associates (The Oily Press Ltd), who retain the copyright to the original article).
In This Section
- Solid-phase extraction columns in the analysis of lipids
- Preparation of Ester Derivatives of Fatty Acids for Chromatographic Analysis
- Preparation of Lipid Extracts Tissues
- The Chromatographic Resolution of Chiral Lipids
- Detectors for HPLC of Lipids with Special Reference to Evaporative Lght-Scattering Detection
- Why Doesn't Your Method Work When I Try It?
- Laboratory Accreditation in a Lipid Analysis Context
- What Column do I Need for Gas Chromatographic Analysis of Fatty Acids?
- Fatty Acid Analysis by HPLC
- Alternatives to Methyl Esters for GC Analysis of Fatty Acids
- A Practical Guide to the Analysis of Conjugated Linoleic Acid (CLA)
- Application of Infrared Spectroscopy to the Rapid Determination of Total Saturated, trans, Monounsaturated, and Polyunsaturated Fatty Acids
- The Use of Lithiated Adducts for Structural Analysis of Acylglycerols by Mass Spectrometry with Electrospray Ionization
- Identification of FAME Double Bond Location by Covalent Adduct Chemical Ionization (CACI) Tandem Mass Spectrometry
- The Use of Countercurrent Chromatography (CCC) in Lipid Analysis
- Gas Chromatographic Analysis of Plant Sterols
- Analysis of Tocopherols and Tocotrienols by HPLC
- Reversed-Phase HPLC of Triacylglycerols
- Structural Analysis of Triacylglycerols
- Thin-Layer Chromatography of Lipids
- High-temperature Gas Chromatography of Triacylglycerols
- Modification of an AOCS Official Method for Crude Oil Content in Distillers Grains and Other Agricultural Materials
- Lipidomics - A Personal View