Metabolism and Function of Very-Long-Chain Polyunsaturated Fatty Acids (>C24) in Mammals
The Authors: Morgan Murray, Chelsey Walchuk and Miyoung Suh.
Introduction
A substantial amount of research has been conducted on the role of long-chain polyunsaturated fatty acids (LC-PUFAs), while interest in very-long-chain polyunsaturated fatty acids (VLC-PUFAs) has emerged due to their rare abundance and highly specialized roles within the tissues of the retina, brain, testis and spermatozoa. VLC-PUFAs can be defined as fatty acids with a chain length greater than 24 carbons and 3 to 6 double bonds. The structures of these fatty acids are unlike other fatty acids as the proximal carboxylic region is comprised of 12 to 20 saturated carbon chains, and the distal region (methyl group end) contains 4 or more conjugated methylene cis double bonds, allowing them to behave like saturated fatty acids in the mobile phase [3]. This unique structure allows these VLC-PUFAs to transcend both the hydrophobic and hydrophilic parts of the lipid bilayer or can be associated with membrane proteins, aiding in their diverse function. Information on the metabolic synthesis and function of VLC-PUFAs is limited and remains to be elucidated. This review will discuss the current knowledge of VLC-PUFAs with regard to their distribution, abundance, biosynthesis and speculated function within the tissues where they are found. In addition, the role of VLC-PUFAs in disease will be addressed including peroxisomal and retinal diseases.
Tissue Distribution and Abundance of VLC-PUFA
Research has shown VLC-PUFAs are isolated within the mammalian body to retinal tissue, testes, brain, and spermatozoa. VLC-PUFAs, unlike LC-PUFAs, cannot be obtained from ordinary dietary sources and thus must be synthesized in situ from shorter fatty acid precursors, to be discussed in further detail shortly. VLC-PUFAs do not exist as free fatty acids in tissues, but rather have their carboxyl head groups esterified to glycerol, forming glycerolipids, phospholipids, or are modified via an amide bond linkage to form sphingolipids and ceramides [3]. Although there are varying chain lengths of VLC-PUFAs, only some of them appear in each of the specialized tissues, indicating tissue specificity.
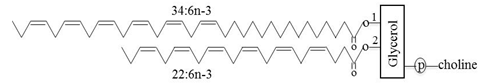
Retinal glycerophospholipids, especially those of photoreceptor membranes, occur as a dipolyunsaturated phosphatidylcholine molecule, with docosahexaenoic acid (DHA, 22:6n-3) as the predominant fatty acid at the sn-2 position and a VLC-PUFA occupying the sn-1 position (Fig. 1). The metabolic process whereby VLC-PUFAs occupy the sn-1 position of phosphatidylcholine is not known. Photoreceptor outer segment membranes, responsible for phototransduction, are highly enriched with VLC-PUFAs, constituting 13% (w/w) of the phosphatidylcholine fraction [4, 18], which is 2 to 3.5 times more than its concentration in the entire retina. The whole eye contains 2-3% (w/w) of VLC-PUFAs in wild type and in fat-1 mice [26]. Current research has indicated that dipolyunsaturated phosphatidylcholines account for approximately 33% of total membrane lipids in the bovine retina [4].
Figure 1. Structure of a phosphatidylcholine molecule with fatty acyl residues of C34:6n-3 and C22:6n-3 in the sn-1 and sn-2 positions, respectively.
Testis- and spermatozoa-VLC-PUFAs are found in sphingomyelin and ceramides, in contrast to the VLC-PUFAs found in the retina [8]. 28:4n-6 and 30:5n-6 fatty acids are the major VLC-PUFAs found in ceramides and sphingomyelin in rat testes [8]. These fatty acids account for 15% (w/w) of the total sphingomyelin fatty acids in testes and spermatozoa during spermatogenesis and 40% (w/w) of total fatty acids in ceramides [17]. Additionally, VLC-PUFAs are found in significantly higher concentrations in sperm heads compared to sperm tails in both bulls and rats [8]. It is clear that interspecies concentrations and structural differences exist as ram and bull spermatozoa contain both n-6 and n-3 VLC-PUFAs, but human and boar spermatozoa are more greatly enriched with n-6 VLC-PUFAs [17]. However, it should be noted that human spermatozoa are enriched with DHA [13] and a further detailed analysis is required for a complete understanding of the full n-6 and n-3 fatty acid profile of spermatozoa. Human spermatozoa also vary in comparison to other mammalian species as they contains 3-4 double bonds up to C32 n-6 fatty acids, while ram, bull and boar spermatozoa contain 5-6 double bonds up to C34 n-6 fatty acids [17].
Brain VLC-PUFAs are similar to retinal fatty acids as they are esterified at the sn-1 position of the glycerol backbone of phosphatidylcholine and contain a saturated, monounsaturated or LC-PUFA at the sn-2 position. The 34:4n-6 and 34:5n-6 fatty acids are the most abundant VLC-PUFAs in the human brain [23]. During development, the concentration of VLC-PUFAs increases in young brains and subsequently declines during older age; a similar trend can be seen in retinal and testicular tissues [17]. These developmental changes may be the result of specific fatty acid incorporation into myelin lipids [3].
Biosynthesis of VLC-PUFA
Numerous studies have confirmed and substantiated the evidence that VLC-PUFAs are synthesized through a series of elongation steps from shorter-chain PUFA precursors in the retina, brain, testis and spermatozoa [29]. Rotstein and Aveldano [20] were the first to report the ability of the retina to synthesize VLC-PUFAs. Through the use of [1-14C] acetate-radiolabeled precursors of LC-PUFAs and VLC-PUFAs or acetate or glycerol, Rotstein and Aveldano [20] confirmed the presence of the necessary machinery within the retina for the biosynthesis of LC- and VLC-PUFAs. It is now known that only those cells, which express the enzyme responsible for the condensation reaction (ELOngase of Very Long chain fatty acid-4, ELOVL4), required for the synthesis of VLC-PUFAs from shorter essential PUFA precursors, produce VLC-PUFAs in situ.
Biosynthesis of VLC-PUFAs is substantially different from synthesis of LC-PUFAs, such as arachidonic acid (AA, 20:4n-6) and DHA, which are synthesized in the liver from essential fatty acids obtained from dietary sources, linoleic acid (LA, 18:2n-6) and α-linolenic acid (α-LNA, 18:3n-3) and are subsequently transported to target tissues [3]. Rather, VLC-PUFAs are synthesized in the specific tissues through a series of elongation and desaturation steps from available shorter-chain PUFAs including LA, α-LNA, eicosapentaenoic acid (EPA, 20:5n-3), docosapentaenoic acid (DPA, 22:5n-3), DHA and AA [24,25]. From these PUFA precursors, C24 fatty acids are generated through the action of seven elongase and fatty acid desaturase enzymes (Fig. 2) [2,25,29]. Elongation and desaturation steps required to produce VLC-PUFAs take place in the endoplasmic reticulum by a membrane-bound, multienzyme complex called the elongase complex [3].
Figure 2. Schematic in vivo biosynthetic pathways of VLC-PUFAs from 18:2n-6 and 18:3n-3 mediated by proteins from the ELOVL family. Consecutive desaturase and elongation steps are performed by Δ5 desaturase, Δ6 desaturase, and ELOVL1-5. * Retroconversion route; **, potential Δ6 desaturase involvement to give rise to n-6 penta- and n-3 hexaenoic VLC-PUFA.
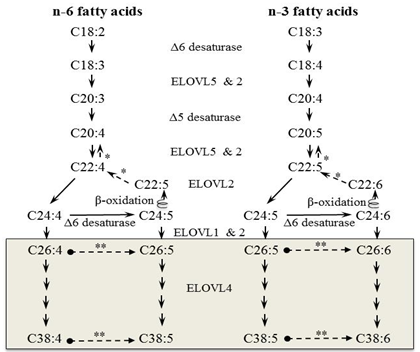
n-3 PUFAs (α-LNA, EPA, DPA and DHA) can be obtained from numerous sources in the diet such as marine fish and flaxseed oil. Alternatively, EPA, DPA and DHA can be biosynthesized from α-LNA. The biosynthesis route appears to be a minor source in adults as a large amount of α-LNA was required in order to raise plasma EPA and DHA levels [15]. Interestingly, n-3 VLC-PUFAs are predominantly synthesized from EPA but not DHA in the rat retina despite having a high concentration of DHA in this tissue [25]. Later studies confirmed this finding in ELOVL4 transduced cell culture models that EPA preferentially elongates to VLC-PUFA over DHA [2,28].
ELOVL4, a condensing enzyme, is currently the only known elongase responsible for the synthesis of VLC fatty acids. This ELOVL4 elongase attaches two-carbon units to the acyl backbone of the fatty acids to produce a longer chain [15]. As previously mentioned, only those tissues that express the ELOVL4 enzyme will produce VLC-PUFAs. The ELOVL4 enzyme is highly expressed in photoreceptor cells of the retina and to a lesser extent in the brain, testis and skin [2]. VLC-PUFAs are not found in the liver or plasma, as these tissues do not express the ELOVL4 protein [29]. Within the adult mouse retina, ELOVL4 mRNA and protein, can be found localized within the inner segments of rod and cone cells [11]. The major VLC-PUFA end products in cells expressing ELOVL4 are 32:5n-3 and 34:5n-3 [2].
Four enzymes and fatty acyl-CoA intermediates carry out the elongation pathway of fatty acyl-CoA. Each round of elongation includes four steps (Fig. 3). Step 1 is a condensation reaction carried out by ELOVL proteins and is generally accepted to be the rate-limiting step of fatty acid elongation. Malonyl-CoA and acyl-CoA are condensed to form the intermediate, β-ketoacyl-CoA. β-Ketoacyl-CoA is then reduced to β-hydroxyacyl-CoA via action of β-ketoacyl reductase and NAD(P)H to conclude step 2. Step 3 results in dehydration by β-hydroxyacyl-CoA dehydratase to yield trans-2,3-acyl-CoA. The final step 4 generates the elongated fatty acyl(n+2)-CoA by reducing enoyl-CoA by enoyl-CoA reductase in the presence of NAD(P)H. The resulting elongated fatty acyl-CoA may then either undergo another round of elongation or be released for use in the cell [3,10].
Figure 3. Fatty acid elongation and β-oxidation pathways. Each round of elongation involves four successive steps in the endoplasmic reticulum. The resulting elongated fatty acyl-CoA product may then either undergo a subsequent round of elongation, be released for use in the cell, or, β-oxidized in the peroxisome. The synthesis involves successive steps of elongation: 1) condensation reaction involving enzyme ELOVL, 2) reduction reaction via β-ketoacyl-CoA reductase, 3) dehydration reaction via β-hydroxyacyl-CoA dehydratase, 4) reduction reaction via enoyl-CoA reductase. β-Oxidation also proceeds via successive steps: 1) oxidation via acylCoA oxidase, 2) and 3) hydration and dehydrogenation via bifunctional proteins, enoyl CoA hydratase and 3-hydroxyacyl CoA dehydrogenase, respectively, 4) thiolytic cleavage via 3-ketoacyl CoA thiolase.
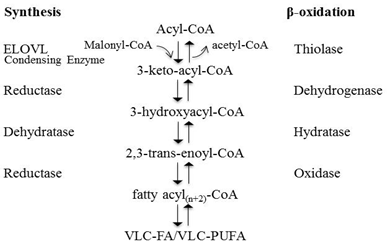
Once synthesized, VLC-PUFAs can be degraded to shorter-chain fatty acids through the β-oxidation pathway, which occurs in peroxisomes (Fig. 3). VLC-PUFAs must first be activated to VLC acyl-CoAs prior to being transported to the peroxisome via the ABC transporter, ABCD1 [10]. The β-oxidation pathway consists of four reactions per cycle: dehydrogenation, hydration, dehydrogenation and thiolytic cleavage [10]. The first dehydrogenation step is catalyzed by peroxisomal straight-chain acyl-CoA oxygenase, followed by hydration and dehydrogenation steps, both of which are catalyzed by the bifunctional enzyme D-bifunctional protein [10]. Lastly, 3-ketoacyl-CoA thiolase carries out the thiolytic cleavage reaction [10]. The products of the β-oxidation pathway may proceed to undergo another cycle of β-oxidation or may undergo other conversions, such as hydrolase or transferase reactions [27].
Physiological Roles of VLC-PUFA
The presence of VLC-PUFAs is unique to the retina, brain, testes and spermatozoa. However, their function is yet to be fully elucidated. Research into peroxisomal and retinal diseases associated with the biosynthesis or β-oxidation of VLC-PUFAs has provided insight into some of the potential roles of VLC-PUFAs in these specific tissues.
Although the function of VLC-PUFAs in brain physiology is not well known, research has demonstrated an increase in the concentrations of VLC-PUFA sphingomyelin during developmental stages, and another change in these concentrations with aging [3]. Furthermore, in individuals with Zellweger syndrome (discussed later), an autosomal recessive inherited disorder associated with a deficiency in peroxisomes and a reduction in the concentration of DHA, VLC-PUFA accumulation occurs in the brain [6]. This accumulation leads to a number of developmental defects and neurodegeneration [6]. These associations of VLC-PUFAs with brain development and Zellweger syndrome suggest that VLC-PUFAs do play an important role in brain physiology.
VLC-PUFAs in the brain may also be related to development as the concentrations of VLC-PUFAs vary during developmental stages compared to the fully developed brain. For example, concentrations of C24 to C38 VLC-PUFAs in brain sphingomyelin are substantially increased during development [3,23]. Furthermore, the concentrations of VLC-PUFAs in the brain tend to be greater in young individuals, including high concentrations of 34:4n-6 and 34:5n-6 [23]. In aging individuals, the overall concentrations of VLC-PUFAs decrease and is replaced with 36:4n-6 as the predominant VLC-PUFA [18,23], suggesting that age-related changes associated with the brain may be linked to the changing concentrations of VLC-PUFAs. However, this remains contentious.
In mammalian testis, VLC-PUFAs are present bound to sphingomyelin and fucosylated gangliosides in germ cells and spermatozoa, however their functions on these lipids have not been determined [10]. Current research has suggested that VLC-PUFAs in the testes are required for maintenance of spermatozoa membranes and spermatogenesis as VLC-PUFA sphingolipids are present during the meiotic division of spermatocytes into spermatids, and are highly enriched in the heads of spermatozoa as VLC-PUFA sphingomyelin [10,19]. Glycosphingolipids and VLC-PUFAs are predicted to be important in spermatozoa development through the stabilization of intercellular bridges between spermatids, the elongation of spermatids through polarization, and stabilizing contact between the spermatid and Sertoli cells [29]. It has also been demonstrated that the concentrations of VLC-PUFAs are correlated with the degree of progression of spermatogenesis and sperm maturation [22]. An example of this correlation was presented in ELOVL2 ablated mice, which is the gene required for the biosynthesis of 24:5n-6 and 30:5n-6 VLC-PUFAs [22]. The inability of these mice to form these VLC-PUFAs led to a halt in spermatogenesis, with the presence of only spermatogonia and primary spermatocytes in the seminiferous tubules [22]. Furthermore, research suggests the importance of VLC-PUFAs in sperm capacitation, a biochemical process required to identify the spermatozoa as competent for oocyte fertilization [22]. Similarly, the link between VLC-PUFAs and spermatogenesis was demonstrated by another study analyzing the fatty acid concentrations of mammalian (bull, cat, rabbit, mouse and rat) testis and the comparison of testicular ceramide and sphingomyelin [8]. Results of this study demonstrated that the presence of VLC-PUFAs in ceramide and sphingomyelin was nonexistent in rats that had not yet reached sexual maturity. At the commencement of spermatogenesis, VLC-PUFAs were present in ceramide and sphingomyelin and these concentrations remained stable throughout fertile adult years of life. At the onset of old age and declining spermatogenesis, VLC-PUFA concentrations declined in rats. Additionally, experimentally induced death of spermatogenic germ cells was implemented through methods such as cryptorchidism, post-ischemic reperfusion, and x-ray irradiation, all of which lead to a reduction in the testicular concentrations of VLC-PUFAs. These very important findings further support the suggestion that VLC-PUFAs are required for the optimal functioning of spermatogenesis and male fertility [8,10,22].
Similar to the brain and testes, the exact function of VLC-PUFAs in the retina is essentially unknown. However, there have been numerous findings related to Stargardt macular dystrophy type 3 (STG3) (described later), an autosomal dominant genetic disorder that has lead researchers to believe that VLC-PUFAs play an integral role in retinal physiology and normal visual functioning. Individuals with STG3 are deficient in VLC-PUFAs due to a mutation on the ELOVL4 gene, required for the biosynthesis of VLC-PUFAs [11,15]. The ELOVL4 protein is highly concentrated in the retina, specifically in the outer segments of photoreceptor cells [3]. Considering STG3 is associated with macular dystrophy and loss of central vision, these findings suggest VLC-PUFAs must be required for optimal visual functioning, specifically in the macular region and photoreceptor cells of the retina.
It has been suggested that the role of VLC-PUFAs in the retina is largely due to the effect of VLC-PUFAs on membrane structure and function [24,26]. The additional 12 to 20 carbons associated with VLC-PUFAs compared to LC-PUFAs have the potential to provide highly distinct roles in membrane structure and fluidity behaving like unusual saturated fatty acids. VLC-PUFAs may allow for distinct membrane fluidity and packing density, creating a unique lipid bilayer that is potentially beneficial for the ability of rod and cone cells to fold back on themselves, aid in the shedding of photoreceptor outer segments, and to stabilize the curved membranes of rod and cone cells [3].
Research has demonstrated a change in the fatty acid concentration of the retina with age, which may have implications for age-related retinal diseases such as age-related macular degeneration (AMD). In the aging eye, a reduction in the concentration of retinal VLC-PUFAs and a reduction in the degree of unsaturation have been demonstrated [21]. The effect of aging and AMD on LC-PUFA and VLC-PUFA concentrations in the retina, retinal pigment epithelium and choroid in human donor eyes has been studied [12]. In the eyes of aging individuals, a substantial decrease in VLC-PUFA concentrations in the retina, retinal pigment epithelium and choroid were demonstrated [12]. These concentrations were further reduced in AMD donor eyes, with a significant reduction in retinal VLC-PUFAs and undetectable concentrations in the retinal pigment epithelium and choroid [12]. These findings may have implications for the use of VLC-PUFAs for the prevention of age-related retinal diseases such as AMD.
To date, research has only just begun to attain information concerning the full biological functions of VLC-PUFAs. Further research will allow for exciting new breakthroughs in determining the roles of these unique fatty acids and their functions to aid in the prevention and maintenance of VLC-PUFA related diseases.
Diseases Associated with VLC-PUFA
Peroxisomal Diseases
Cerebro-hepato-renal syndrome or Zellweger syndrome, X-linked adrenoleukodystrophy, acyl-CoA oxidase1 deficiency and D-bifunctional protein deficiency belong to a group of peroxisomal biogenesis disorders often referred to as the Zellweger spectrum [1,6,10]. Zellweger syndrome is an inherited autosomal recessive disorder caused by a mutation of the PEX gene responsible for the encoding of peroxins, which are required for normal biological functioning of the peroxisomes [6]. As previously described, peroxisomes are required for the β-oxidation of VLC-PUFAs. Considering the peroxisomal deficiency associated with Zellweger syndrome a toxic accumulation of VLC-PUFAs in the brain occurs in these patients, along with a deficiency of DHA and an increase in fatty acid saturation [23]. The predominant VLC-PUFAs present in Zellweger brains are penta- and hexaenoic acids, whereas a normal brain contains C32 to C38 tetra- and pentaenoic acids [23]. Zellweger brains also contained trace amounts of C40 n-6 VLC-PUFAs, which are absent in normal brains [3]. Clinical symptoms associated with this disease are highly severe and include psychomotor delay, dysmorphia, neonatal seizures, retinopathy, cataracts and hearing loss [6].
X-Linked andrenoleukodystrophy is the most common peroxisomal disorder (1 in 17,000 births) and is associated with a mutation in the ABCD1 gene required for β-oxidation [7,10]. The ABCD1 transporter is required for the transport of VLC acyl-CoAs to peroxisomes for further degradation to long-chain fatty acids [10]. In individuals with X-linked andrenoleukodystrophy, this leads to an accumulation of VLC-PUFAs and a number of related symptoms such as developmental delays and demyelination of the central nervous system [10]. Two other peroxisomal biogenesis disorders include a deficiency of acyl-coA oxidase and D-bifunctional protein [10]. Acyl-CoA oxidase is required to catalyze the first step of β-oxidation, the dehydrogenation reaction. A deficiency of this enzyme creates symptoms such as retinopathy, hypotonia and delayed motor development [10]. D-bifunctional protein is required for the hydration and dehydrogenation reactions of β-oxidation. A deficiency in D-bifunctional protein leads to developmental delays, craniofacial dysmorphia, neonatal seizures and hepatomegaly [10].
It has been determined that individuals with peroxisomal biogenesis disorders such as Zellweger syndrome are highly deficient in DHA in the brain (as low as 20% of normal DHA values) with even lower concentrations in the retina [14]. The dangerously low concentrations of DHA in the brain and retina may be associated with the majority of severe symptoms of Zellweger syndrome [10]. Abe et al. [1] determined that DHA-containing phospholipid (phosphatidylcholine, phosphatidylethanolamine, plasmenylethanolamine) levels were significantly reduced in individuals with peroxisomal biogenesis disorders including peroxisome acyl-coA oxidase1 (reduced by 50%) and D-bifunctional protein deficient fibroblasts, but these levels were unchanged in individuals with X-linked andrenoleukodystrophy.
It has also been demonstrated that in skin fibroblasts from patients with Zellweger syndrome and acyl-coA oxidase deficiency, VLC-saturated fatty acids, VLC-monounsaturated fatty acids and VLC-PUFAs were accumulated in phosphatidylcholine but not in individuals with X-linked andrenoleukodystrophy [1]. Phosphatidylcholine molecules in skin fibroblasts included saturated, monounsaturated, and diunsaturated VLC-fatty acids including, 42:1, 42:2, 44:0, and 44:1 [1], with 26:0 acylated at the sn-1 position and 16:0 at the sn-2 position of the glycerol backbone [1,9]. LC-PUFAs and VLC-PUFAs including 24:4, 24:5 and 26:5 were elevated in phosphatidylcholine 42:5 of skin fibroblasts from patients with Zellweger syndrome and acyl-CoA oxidase deficiency [1]. In brains of individuals with peroxisomal biogenesis disorders including Zellweger syndrome and andrenoleukodystrophy, an accumulation of phosphatidylcholine containing VLC-PUFAs from C32 to C40 was found along with an increase in unsaturation [23]. Considering the varied levels of fatty acid accumulation within these disorders, it is most likely a combination of toxic levels of VLC-PUFAs and a deficiency in DHA that leads to the number of severe symptoms associated with Zellweger spectrum related diseases.
Retinal Diseases: Stargardt Macular Dystrophy Type 3 (STG3)
Stargardt macular dystrophy (STG) is a genetic disorder present in 1 out of 10,000 individuals [16]. STG is present in two forms including an autosomal recessive and autosomal dominant form [16]. A very rare form of STG is STG3, an autosomal dominant genetic disorder associated with juvenile macular dystrophy [16]. STG3 is caused by three independent mutations of the ELOVL4 gene leading to a frame shift and the incorporation of a premature stop codon, resulting in the loss of the C-terminal region of the protein [10,16]. The truncated ELOVL4 proteins result in mislocalization and a loss in enzymatic abilities [10]. Considering that ELOVL4 proteins are required for the production of VLC-PUFAs, individuals with STG3 lack the ability to fully produce these fatty acids. As previously mentioned, the role of VLC-PUFAs in the retina is not fully understood, but it has been suggested that the deficiency in VLC-PUFAs leads to damage of the outer segments of photoreceptors affecting phototransduction, leading to the accumulation of lipofuscin and eventual photoreceptor cell death [5].
Furthermore, a deficiency in VLC-PUFAs in these individuals has led to a number of vision-related symptoms that appear during adolescence, including the deterioration of the retinal pigment epithelium and photoreceptor cells and accumulation of lipofuscin leading to a loss of visual acuity and central vision [16]. The natural course of VLC-PUFA concentrations in this disease was demonstrated in ELOVL4 transgenic mice (ELOVL4/TG1-2 line) by electroretinography, fundus imaging and fatty acid analysis [11]. The decrease of VLC-PUFAs in the retina was not demonstrated until 3 months of age and continued until 24 months of age with no changes in retinal function prior to this time. At two months of age, degradation of rod photoreceptors was determined through fundus photography, with a complete disappearance of rod photoreceptors by 18 months. No changes in cone photoreceptors were demonstrated until 15 months of age, with a very limited number of cone cells present by 24 months of age, which shows the ELOVL4 mutation affects cones much later than rods in this mouse model. However, although there was a decline in VLC-PUFAs from 3 to 24 months of age, these fatty acids were still being synthesized in lesser amounts with peak concentrations at 6 to 9 months of age. Kuny et al. [11] suggest that although it is clear that VLC-PUFAs play a role in optimal retinal functioning, there may be other factors leading to the degradation of photoreceptor cells in individuals with STG3.
Conclusion
This review provides insight into the integral role of VLC-PUFAs in retinal, testicular, spermatozoa and cerebral tissues. Although their full function and metabolism remain to be elucidated, substantial research exists highlighting disease states resulting from VLC-PUFA dysfunction. To date, research has only just begun to attain information concerning the full biological functions of VLC-PUFAs. Further research is warranted to determine the roles and distribution of these novel fatty acids, as well as their functions, possessing the capability to aid in the prevention and maintenance of VLC-PUFA related diseases.
Abbreviations: AA, arachidonic acid; ABCD1, ABC transporter; AMD, age-related macular degeneration; DHA, docosahexaenoic acid (22:6n-3); DPA, docosapentaenoic acid (22:5n-3 and 22:5n-6); ELOVL, Elongase of very long chain fatty acid; EPA, eicosapentaenoic acid (20:5n-3); LA, linoleic acid (18:2n-6); LC-PUFA, long-chain polyunsaturated fatty acids; LNA, α-linolenic acid (18:3n-3); NADPH, nicotinamide adenine dinucleotide phosphate; STG3, Stargardt disease type 3; VLC-PUFA, very-long-chain polyunsaturated fatty acids.
References
- Abe, Y., Honsho, M., Nakanishi, H., Taguchi, R. and Fujiki, Y. Very-long-chain polyunsaturated fatty acids accumulate in phosphatidylcholine of fibroblasts from patients with zellweger syndrome and acyl-CoA oxidase1 deficiency. Biochem. Biophys. Acta, 1841, 610-9 (2014) (DOI: 10.1016/j.bbalip.2014.01.001).
- Agbaga, M.P., Brush, R.S., Mandal, M.N.A., Henry, K., Elliott, M.H. and Anderson, R.E. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc. Natl. Acad. Sci. U.S.A., 105, 12843-12848 (2008) (DOI: 10.1073/pnas.0802607105).
- Agbaga, M.P., Mandal, M.N.A. and Anderson, R.E. Retinal very long-chain PUFAs: new insights from studies on ELOVL4 protein. J. Lipid. Res., 51, 1624-1642 (2010) (DOI: 10.1194/jlr.R005025).
- Alvedaño, M.I. A novel group of very long chain polyenoic fatty acids in dipolyunsaturated phosphatidylcholines from vertebrate retina. J. Biol. Chem., 262, 1172-1179 (1987).
- Berdeaux, O. and Acar, N. Very-long-chain polyunsaturated fatty acids in the retina: analysis and clinical relevance in physiological and pathological conditions. OCL., 18, 284-290 (2011) (DOI: 10.1051/ocl.2011.0406).
- Crane, D.I. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem. Int., 69C, 1-8 (2014) (DOI: 10.1016/j.neuint.2014.02.007).
- Engelen, M., Kemp, S., de Visser, M., van Geel, B.M., Wanders, R.J.A., Aubourg, P. and Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis., 7, 1172-7-51 (2012) (DOI: 10.1186/1750-1172-7-51).
- Furland, N.E., Zanetti, S.R., Oresti, G.M., Maldonado, E.N. and Aveldano, M.I. Ceramides and sphingomyelins with high proportions of very long-chain polyunsaturated fatty acids in mammalian germ cells. J. Biol. Chem., 282, 18141-18150 (2007) (DOI: 10.1074/jbc.M700708200).
- Hama, K., Nagai, T., Nishizawa, C., Ikeda, K., Morita, M., Satoh, N., Nakanishi, H., Imanaka, T., Shimozawa, N., Taguchi, R., Inoue, K. and Yokoyama, K. Molecular species of phospholipids with very long chain fatty acids in skin fibroblasts of Zellweger syndrome. Lipids, 48, 1253-1267 (2013) (DOI: 10.1007/s11745-013-3848-5).
- Kihara, A. Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem., 152, 387-95 (2012) (DOI: 10.1093/jb/mvs105).
- Kuny, S., Filion, M.A., Suh, M., Gaillard, M. and Sauvé, Y. Long-term retinal cone survival and delayed alteration of the cone mosaic in a transgenic mouse model of Stargardt-like dystrophy (STGD3). Invest. Ophthalmol. Vis. Sci., 55, 424-439 (2014) (DOI: 10.1167/iovs.13-13457 ).
- Liu, A., Chang, J., Lin, Y., Shen, Z. and Bernstein, P.S. Long-chain and very long-chain polyunsaturated fatty acids in ocular aging and age-related macular degeneration. J. Lipid. Res., 51, 3217-3229 (2010) (DOI: 10.1194/jlr.M007518).
- Martinez, J.C., Landeras, J. and Gadea, J. Spermatozoa and seminal plasma fatty acids as predictors of cryopreservation success. Andrology, 1, 365-375 (2013) (DOI: 10.1111/j.2047-2927.2012.00040.x).
- Martinez, M. Polyunsaturated fatty acids in the developing human brain, erythrocytes and plasma in peroxisomal disease: therapeutic implications. J. Inherit. Metab. Dis.,18, Suppl. 1, 61-75 (1995) (DOI: 10.1007/BF00711429).
- McMahon, A. and Kedzierski, W. Polyunsaturated very-long-chain C28-C36 fatty acids and retinal physiology. Br. J. Opthalmol., 94, 1127-1132 (2010) (DOI: 10.1136/bjo.2008.149286).
- Molday, R.S. and Zhang, K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog. Lipid Res., 49, 476-92 (2010) (DOI: 10.1016/j.plipres.2010.07.002).
- Poulos, A., Sharp, P., Johnson, D., White, I. and Fellenberg, A. The occurrence of polyenoic fatty acids with greater than 22 carbon atoms in mammalian spermatozoa. Biochem. J., 240, 891-895 (1986).
- Poulos, A. Very long chain fatty acids in higher animals - a review. Lipids, 30, 1-14 (1995) (DOI: 10.1007/BF02537036).
- Robinson, B.S., Johnson, D.W. and Poulos, A. Novel molecular species of sphingomyelin containing 2-hydroxylated polyenoic very-long-chain fatty acids in mammalian testes and spermatozoa. J. Biol. Chem., 267, 1746-51 (1992).
- Rotstein, N.P. and Aveldaño, M.I. Synthesis of very long chain (up to 36 carbon) tetra, penta and hexaenoic fatty acids in retina. Biochem. J., 249, 191-200 (1988).
- Rotstein N.P., Ilincheta de Boschero, M.G., Giusto, N.M. and Aveldano, M.I. Effects of aging on the composition and metabolism of docosahexaenoate-containing lipids of retina. Lipids, 22, 253–260 (1987) (DOI: 10.1007/BF02533988).
- Sandhoff, R. Very long chain sphingolipids: tissue expression, function and synthesis. FEBS Lett., 584, 1907-1913 (2010) (DOI: 10.1016/j.febslet.2009.12.032).
- Sharp, P., Johnson, D. and Poulos, A. Molecular species of phosphatidylcholine containing very long chain fatty acids in human brain: Enrichment in X-linked adrenoleukodystrophy brain and diseases of peroxisome biogenesis brain. J. Neurochem., 56, 30-7 (1991) (DOI: 10.1111/j.1471-4159.1991.tb02558.x).
- Suh, M., Wierzbicki, A.A., Lien, E.L. and Clandinin, M.T. Dietary 20:4n-6 and 22:6n-3 modulates the profile of long- and very-long-chain fatty acids, rhodopsin content, and kinetics in developing photoreceptor cells. Ped. Res., 48, 524-530 (2000) (DOI: 10.1203/00006450-200010000-00017).
- Suh, M. and Clandinin, M.T. 20:5n-3 but not 22:6n-3 is a preferred substrate for synthesis of n-3 very-long-chain fatty acids (C24-C36) in retina. Curr. Eye Res., 30, 959-968 (2005) (DOI: 10.1080/02713680500246957).
- Suh, M., Sauvé, Y., Merrells, K.J., Kang, J.X. and Ma, D.W.L. Supranormal electroretinogram in Fat-1 mice with retinas enriched in docosahexaenoic acid and n-3 very long chain fatty acids (C24-C36). Invest. Ophthalmol. Vis. Sci., 50, 4394-4401 (2009) (DOI: 10.1167/iovs.08-2565).
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res., 51, 2863-2895 (2010) (DOI: 10.1194/jlr.R005959).
- Yu, M., Benham, A., Logan, S., Brush, R.S., Mandal, M.N.A., Anderson, R.E. and Agbaga, M.P. ELOVL4 protein preferentially elongates 20:5n3 to very long chain PUFAs over 20:4n6 and 22:6n3. J. Lipid Res., 53, 494-504 (2012) (DOI: 10.1194/jlr.M021386).
- Zadravec, D., Tvrdik, P., Guillou, H., Haslam, R., Kobayashi, T., Napier, J.A., Capecchi, M.R. and Jacobsson, A. ELOVL2 controls the level of n-6 28:5 and 30:5 fatty acids in testis, a prerequisite for male fertility and sperm maturation in mice. J. Lipid Res., 52, 245-55 (2011) (DOI: 10.1194/jlr.M011346).