Detectors for HPLC of Lipids with Special Reference to Evaporative Lght-Scattering Detection
The Author: William W. Christie, James Hutton Institute (and Mylnefield Lipid Analysis), Invergowrie, Dundee (DD2 5DA), Scotland.
The following was first published by W.W. Christie in Advances in Lipid Methodology - One, pp. 239-271 (1992) (Ed. W.W. Christie, Oily Press, Ayr), and it is reproduced here by kind permission of P.J. Barnes & Associates (The Oily Press). This review is now out of date in terms of applications and chromatographic media, and especially in regard to mass spectrometry, but no comparable review has been published recently and it remains a useful guide to the early literature. The author's book (written jointly with Xianlin Han) Lipid Analysis (4th edition) (Oily Press, Bridgwater) brings mass spectrometric detection up to date. A comprehensive list of recent references to the topic can be accessed here. The charged aerosol detector is a variant on the evaporative light-scattering detector, which has become available too recently for discussion here (Moreau, R.A. Lipid analysis via HPLC with a charged aerosol detector. Lipid Technol., 21, 191-194 (2009); DOI: 10.1002/lite.200900048).
- Introduction
- Optical and Spectrophotometric Detectors
1. Differential refractometry
2. Ultraviolet spectrophotometry
3. Fluorescence detection
4. Infrared spectrophotometric detectors
5. Spectrophotometric detection with post-column chemical reaction - Some Miscellaneous Detection Systems
1. The mass spectrometer as an HPLC detector
2. Radioactivity detectors
3. Density, electrochemical and other detectors - Transport-flame Ionization Detectors
1. Apparatus
2. Applications of transport-flame ionization detectors - Evaporative Light-Scattering Detectors
1. Construction and the nature of the response
2. Applications of light-scattering detection in lipid class separations
3. Applications of light-scattering detection in separations of molecular species of lipids
4. Some miscellaneous separations - References
Introduction
Detectors functioning according to many different principles are available as a means of sensing solutes in the mobile phase as they elute from the column during high-performance liquid chromatography (HPLC) of lipids. Defined peaks may be quantified directly or fractions containing the solutes can be collected for analysis by other means. The topic of detectors for HPLC analysis of lipids was extensively reviewed by the author in 1987 [14] and that work should be be consulted for specific early applications. However, the much wider use of light-scattering detectors in the last few years has changed the perspective greatly. In discussing different detection systems here, the author has been highly selective in his choice of examples, with concentration on more recent papers. This review is to some extent a supplement to the earlier work avoiding unnecessary repetition, but applications of light-scattering detectors are discussed at length. Others have reviewed HPLC separations of lipids in general [59,109,110] and phospholipids and glycolipids in particular [68,69].
As most lipids lack chromophores that permit specific spectrophotometric detection, this most essential process has been the weak link in the chromatographic system. The availability of one particular detector in a laboratory may determine which solvents can be used in the mobile phase, whether they can contain ionic species, whether gradient elution is possible, and sometimes even which mode of chromatography and which stationary phase are appropriate. For example, if a differential refractometer is the only detector to which the analyst has access, a mobile phase of constant composition, i.e. isocratic elution, must be used and some compromise in the quality of the separation may have to be accepted. Most lipid classes are heterogeneous and contain aliphatic moieties with a range of chain lengths and variable numbers of double bonds. Each type of detector has its idiosyncrasies and responds to such structural features of lipids in a different manner, and it must be calibrated with appropriate standards.
For some time, it was thought that detectors working on the transport-flame ionization principle (see Section D below) would be the ideal answer to the problem, but the commercial instruments have been disappointing for a variety of reasons. They certainly have a theoretical potential to exhibit a linear response that may be largely independent of the structure of the solute, and it is to be hoped that this potential will one day be fulfilled. On the other hand, evaporative light-scattering or "mass" detectors (Section E below) are available now, and their virtues are becoming increasingly evident. They are simple, rugged and versatile in use, and afford high sensitivity in comparison to other "universal" detectors. Almost any solvent can be used in the mobile phase in complex gradients. It must be admitted that the response does not bear a simple linear relationship to the amount of solute and varies in a poorly understood manner with the nature of the lipid. Yet the advantages of these detectors far outweigh the disadvantages, and with care it is possible to live with the latter provided that they are properly understood. It is the author's opinion that they offer much more to lipid analysts than any alternative, and there does not appear to be any superior system on the horizon. Further, the analyst now has a choice of instruments from at least four manufacturers.
Optical and Spectrophotometric Detectors
1. Differential refractometry
Refractive index (RI) detectors of three different kinds are in common use, i.e. deflection, Fresnel and interference refractometers. All can be considered as universal in scope and can be used with any solute with a different refractive index from that of the mobile phase. In operation, they monitor the difference in refractive index between the eluent and the pure mobile phase continuously. Although differential refractometers can in theory be used with gradients by having a reference column in parallel with the main one, it is almost impossible to achieve a balance between the two and thence a stable base-line in practice. They are at their best with isocratic elution, although programming of the flow-rate or temperature of the mobile phase affords some limited opportunities to enhance separations. In order to minimize changes in solvent composition and base-line drift during a chromatographic run, volatile solvents should be avoided. When a static reference cell is used, it is often necessary to flush it out with fresh mobile phase at some point during a day's work. Lipid samples should be dissolved in fresh mobile phase for injection onto the HPLC column. RI detectors are very sensitive to fluctuations in temperature, so many commercial instruments have some means of controlling this, varying from water-circulation to a more sophisticated thermostatted system. For this reason, they should be located away from sun-lit windows and from draughts, such as are found near doors or fume cupboards. Other disadvantages are that sample peaks can be both positive and negative, and that bubbles of gas in the solvents, the flow-rate of the mobile phase, leaks in the system, back pressure and pulsations of the HPLC pump can influence base-line stability.
With simple lower-cost RI detectors, solute components amounting to about 10 micrograms can perhaps be detected. A 10 to 30 times improvement in this sensitivity may be possible with precise control of temperature and other chromatography parameters in the best commercial instruments. Comparatively little use has been made of RI detectors for quantitative analysis of lipids, and there has been some debate on whether response factors are necessary for different lipid classes or molecular species [14]. In analyses of molecular species of triacylglycerols at least, the consensus appears to be that acceptable results can be obtained by equating detector response directly with the mass of components, but careful calibration and calculation of response factors will improve accuracy.
Such detectors were once relatively common in lipid laboratories, and have been used especially for preparative-scale chromatography and for gel filtration, but are nowadays used much less. They are at their best for the preparative-scale isolation, under isocratic elution conditions, of particular lipid components that are required for analysis by other procedures. In this way, they have been applied to the isolation of neutral lipids, phospholipids and molecular species of both, and fatty acid derivatives of various kinds [14]. The limits of development of RI detection may have been reached by Frede et al. [28,29], who developed a system involving temperature programming for the separation of molecular species of milk triacylglycerols by HPLC in the reversed-phase mode. Programming of the flow-rate of the mobile phase can also be of assistance with related samples [3].
2. Ultraviolet spectrophotometry
Spectrophotometric detectors in the ultraviolet (UV)-visible range for HPLC are used more frequently than any other by analysts in general, so they are relatively inexpensive and tend to be one of the first to which lipid analysts have access. Detectors constructed specifically for HPLC use with a cell volume of about 8 microlitres are recommended (as opposed to UV spectrophotometers with a flow-cell as an optional extra), and only those affording continuously variable wavelengths are of much value to lipid analysts. UV detectors can sometimes give great selectivity and sometimes sensitivity in the analysis of specific compounds, and they are relatively insensitive to changes in ambient temperature or the flow-rate of the mobile phase. While they can be used in gradient elution applications on occasion, base-line drift can be troublesome. A detector cell can easily become contaminated in use, although this may not be immediately obvious.
Compounds containing conjugated double bond systems and aromatic rings give much the best response, but such functional groups are only rarely found in natural lipids. There are some seed oils containing fatty acids with conjugated double bond systems, and these are often present in lipids subjected to hydroperoxidation through chemical or enzymatic action, as in many biologically important eicosanoids for example [102]. UV detection is the most common procedure used with autoxidized lipids [14]. In addition to mere detection, second-derivative UV spectroscopy has proved of value for the identification of configurational isomers of conjugated dienes formed in oxidized phospholipids [42]. cis,trans- and trans,trans-Dienes were easily distinguished, for example. If lipids are defined in a broad sense, carotenes and tocopherols can be subjected to HPLC analysis with UV detection. Among published applications, modern diode array detectors, which covered a range of wavelengths, were employed to detect and identify cholesterol esters, sterols, dolichol, ubiquinone, alpha-tocopherol and retinol in tissue extracts [32] and acyl-coenzyme A esters [140] after separation by HPLC.
An alternative approach has been to convert lipids to compounds that absorb strongly in the UV range. For example, fatty acids have been esterified with aromatic alcohols for analysis, the carbohydrate moieties of glycolipids have been benzoylated, and diacylglycerols derived from phospholipids have been esterified with aromatic acids. Following conversion to the phenacyl or naphthacyl esters, fatty acids are easily resolved by HPLC in the reversed-phase mode on chemically bonded octadecylsilyl (ODS) or octylsilyl phases for UV detection at 242 to 254 nm [14]. The nature of the separation is dependent both on the chain length of the fatty acid and the number of double bonds, each double bond reducing the retention time by the equivalent of about two methylene groups. Whether this technique has any real analytical value per se is dubious, since gas chromatography (GC) offers much more in terms of convenience and ease of identification of components [17]. Reversed-phase HPLC used in this way certainly does have advantages for the analysis of fatty acids containing functional groups that are thermally labile, e.g. cyclopropene fatty acids [141] and perhaps for those with trans-double bonds [38,127], but otherwise the technique is best considered as a micro-preparative method for the isolation of fatty acids for, say, structural analysis or radioactivity measurements. Phenacyl esters of fatty acids have also been separated by HPLC in the silver ion mode, i.e. by the number and configuration of the double bonds, and this technique may have some potential for the analysis of fatty acids with trans-double bonds [21].
Benzoylation of glycosphingolipids for HPLC analysis has proved its worth in terms of ease of detection by UV spectrophotometry, and by reducing polarity and simplifying resolution, both in the separation of different glycolipid classes and for molecular species of these [14,68]. The response is dependent on the number of benzoyl groups that react in each instance, but can usually be related to the molar amount of the native lipid. Similarly, a well-established technique for the analysis of molecular species of phospholipids consists in hydrolysing them with phospholipase C, followed by conversion to the benzoyl or dinitrophenyl urethane derivatives, for separation by HPLC in the reversed-phase mode for UV detection [14]. The technique has the advantages that the same type of derivative can be made from any phospholipid class, and that detection is independent of the fatty acid compositions of components so molar proportions are determined directly. Naphthylethylcarbamate derivatives have been utilized to facilitate chiral separations. Among recent applications, UV-absorbing derivatives have been employed for the analysis of acylcarnitines [78] and lysogangliosides [57].
With lipids or derivatives of these kinds with which specific wavelengths can be selected, good quantification in direct molar terms is usually possible with UV spectrophotometric detection. Both gradient and isocratic elution methods can be used if the composition of the mobile phase is chosen with care and other chromatographic parameters are carefully controlled and standardized.
Most natural lipids exhibit a weak absorbance in the range 200 to 210 nm of the UV spectrum, sometimes termed "end absorption", that is the result of the presence of isolated double bonds predominantly, although carbonyl, carboxyl, phosphate, amino and quaternary ammonium and other functional moieties make some contribution [49]. When UV detection at such wavelengths is used in lipid analysis, several disadvantages become apparent. Many solvents of proven value in the chromatography of lipids, such as chloroform, acetone, toluene or ethyl acetate, absorb strongly and cannot be used in mobile phases. Others such as ethers require to be carefully purified to eliminate contaminants that absorb in the appropriate region. Similarly, those solvents which are transparent at the required wavelengths, e.g. hexane, isopropanol, acetonitrile, methanol and water, must be of very high purity since traces of extraneous materials with appreciable extinction coefficients, such as antioxidants or plasticisers, could seriously impair base-line stability and give high background values. In samples for analysis, impurities and natural substances, such as peroxidized lipids or tocopherols, can give disproportionately large peaks in the HPLC trace, obscuring the components of interest.
In spite of these difficulties, UV detection at these low wavelengths has been very widely used in lipid analysis, and especially in the separation of simple and phospholipid classes [14]. Even with the restricted range of solvents that can be employed in mobile phases, some versatility remains to change the selectivity in particular analyses. For example in the separation of phospholipids, hexane-isopropanol-water or acetonitrile-methanol-water mixtures are commonly used; the order of elution of the neutral phospholipids is not greatly affected by the type of mobile phase employed, but acidic phospholipids, such as phosphatidylserine and phosphatidylinositol, tend to elute much earlier when acetonitrile is present.
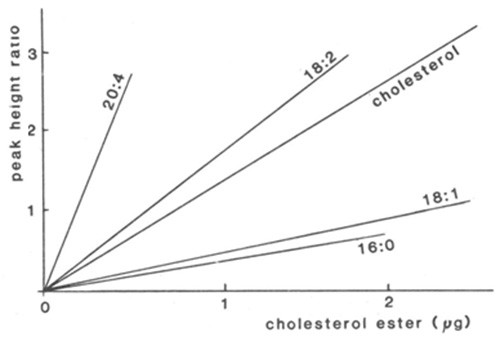
Because variation in the degree of unsaturation of each component can make a substantial difference to the response at low wavelengths, direct quantification is difficult and relatively saturated lipids might even be overlooked. By determining the apparent extinction coefficient for each component in a standard mixture very similar to those to be analysed, direct quantification has been attempted by many analysts. For example, calibration lines for cholesterol and cholesterol esters at 210 nm are shown in Figure 1 [138]. There was a good rectilinear response for each fatty acid, but the slope of the line varied greatly according to degree of unsaturation. This of course means that it is necessary to be certain of the identity of every peak (and these should contain only one species), whenever direct quantification is attempted. This is perhaps an extreme example and smaller correction factors were found to be required in analyses of lipid classes, where the differences in the fatty acid compositions of the separated components were less (c.f. reference [39]). When samples are likely to be variable in composition, it is advisable to collect the peaks as they emerge from the detector for estimation by appropriate micro-methods. Most analysts have followed the latter approach, using phosphorus analysis for phospholipids, for example. GC analysis of the methyl ester derivatives of fatty acid constituents, prepared from fractions, with an added internal standard permits identification and quantification simultaneously and has wider applicability [14,17].
Figure 1. Calibration lines for the quantification of cholesterol esters by HPLC with detection at 210 nm [138]. The ratio of the peak heights of the components to that of cholesteryl heptadecanoate (internal standard) are plotted as a function of the mass of each cholesterol ester standard, i.e. cholesteryl arachidonate (20:4), linoleate (18:2), oleate (18:1) and palmitate (16:0) and for unesterified cholesterol. (Reproduced by kind permission of the authors and of Atherosclerosis, and redrawn from the original paper).
Direct detection (without response factors) and quantification at 215 to 230 nm, where ester bonds exhibit a distinct absorbance but double bonds do not, has been used for analysis of molecular species of triacylglycerols separated by reversed-phase HPLC [40]. With mixtures of acetonitrile and hexane-isopropanol as the mobile phase and detection at 215 nm, as little as 1 microgram of triacylglycerol could be analysed. These results have been confirmed and extended by others [31,111], and there would appear to be no reason why this form of detection should not be extended to other types of sample.
3. Fluorescence detection
Only a few rare lipids exhibit natural fluorescence, but it is possible to make use of the high sensitivity (up to a hundred times greater than absorption detectors) and selectivity of fluorescence detection by preparing suitable derivatives of lipids for chromatography. Detectors of this kind have a wide dynamic range. Unfortunately, the response is affected substantially by the nature of the mobile phase, and careful calibration is necessary when gradient elution conditions are employed [66]. The detector has been used most often in the analysis of fatty acids, separated by reversed-phase HPLC, after conversion to suitable derivatives. Anthrylmethyl esters have been used most often for the purpose, but many different alternatives have been suggested, some offering sensitivities to the femtomole level [14]. At these concentrations, such analytical systems offer real competition to GC in some circumstances, and especially for the analysis of the biologically important free fatty acid fraction in small samples of plasma. The wide variety of lipid classes found in animal tissues but including analogues containing pyrenyl fatty acids, which are detectable to levels of 10-13 mole by their fluorescence, have been separated by HPLC and quantified following correction for quenching by the mobile phase [43]. The ternary gradient elution procedure was based on one developed originally for use with an evaporative light-scattering detector (see Section E.2 below) [12]. Among other recent applications, fluorescence detection has been applied for the analysis of amino-phospholipids (phosphatidylethanolamine, phosphatidylserine and their lyso-forms) [50], phosphatidic acid [142], platelet-activating factor [79] and its lyso-form [108], diacylglycerols derived from phospholipids [104] and monosialogangliosides [144].
4. Infrared spectrophotometric detectors
Infrared (IR) detectors have been used to a limited extent only for the analysis of non-polar lipids, with the specific absorbance for the carbonyl function between 1650 and 1860 cm−1 (or at about 5.75 microns) being the spectral region of value [35]. The detector is not sensitive to variations in ambient temperature or the flow-rate of the mobile phase, but most solvents tend to absorb to some extent at least at the key wavelengths, so causing high background values. A consequence is that marked base-line drift is seen whenever gradient elution is used. Although the effect is diminished in cells with a short path length (1 to 2 mm), the sensitivity is then reduced. Hexane, acetonitrile, chloroform, methylene chloride and tetrahydrofuran appear to be the most useful solvents for IR detection. No quantitative studies have been performed with lipids, but the detection limit was about 1 microgram [35].
Others used a purpose-built infrared detector with a helium-neon laser operating at 3.39 microns, the CH2- stretching frequency, in separations of a limited range of model simple lipids [117]. However, few solvents are permissible in the mobile phase, chloroform being utilized in this application, so that this form of detection has no obvious practical value.
5. Spectrophotometric detection with post-column chemical reaction
The content of particular analytes in an HPLC mobile phase can be monitored by incorporating a reaction chamber after the column, where mixing with specific chemical or enzymatic reagents takes place so that the products can be quantified by spectrophotometric detection. Among the more familiar applications to lipids that have been described are chemical determination of phosphate in phospholipids and enzymatic determination of triacylglycerols and cholesterol in lipoproteins or lipid extracts [14]. While such detection systems can offer high specificity, they are inherently limited in scope.
More recent applications of reaction and post-column detection in the analysis of lipids by HPLC include separations of molecular species of monoacylglycerols [128] and mono-, di- and triacylglycerols [58], fluorescent detection using the probe 1,6-diphenyl-1,3,5-hexatriene for quantification of molecular species of phospholipids [101] and an iron-thiocyanate assay for hydroperoxides [87]. In addition, hydroperoxides have been detected with high specificity and sensitivity (picomolar levels) by making use of the hydroperoxide-dependent chemiluminescence produced by luminol oxidation during the reaction of hydroperoxides with cytochrome C-haeme [46,80-82,145-147]. Poly-phosphoinositides have been detected at low concentrations by means of a novel method of this type involving a "metal-dye" interaction [74,75]. As inositol phosphates emerged from the HPLC column, they encountered an yttrium salt with which they rapidly formed pH-dependent complexes. A "reporter" dye, 4-(2-pyridylazo)resorcinol, was used to sense formation of the complex by producing a negative adsorption peak at 520 to 550 nm. The method was reportedly usable down to picomole levels.
Some Miscellaneous Detection Systems
1. The mass spectrometer as an HPLC detector
Mass spectrometry (MS) is a powerful analytical tool that can supply both structural information about compounds and quantitative data relating to mass. Under optimum conditions, it can provide the molecular weight, the empirical formula and often the complete structure of an unknown compound in addition to giving a measure of the amount present. For some years it was necessary to volatilize the sample in the ion source of the instrument before ionizing it by electron-impact or chemical-ionization techniques, but recently methods have been developed for producing ions from materials in the condensed phase (e.g. by fast-atom bombardment techniques). While combined GC-MS has been available for many years, it has proved more difficult to marry HPLC with MS, because of problems in removing the solvent prior to ionization. Many of the difficulties have now been overcome; several different HPLC-MS interfaces are available on commercial instruments, and the construction and properties of these have been reviewed [51].
One of the first methods of transferring an HPLC solute to the ion source of the mass spectrometer was the field-desorption technique, in which the sample is deposited on a wire emitter in liquid solution, dried and then inserted into the instrument in order to obtain the spectrum. The technique can give good results but is not a continuous one. A second approach to interfacing an HPLC eluent with a mass spectrometer makes use of a mechanical transport system, similar to that used with flame-ionization detection (see Section D below), in which the sample is deposited on a moving belt and the solvent is evaporated before the solute reaches the ion source. A spray deposition technique is usually favoured to promote even deposition and evaporation. In its simplest form, the vaporization-ionization principle is field desorption, but electron-impact and chemical ionization techniques can also be employed. Of greater value is a "thermospray" method in which a supersonic jet of vapour carrying entrained particles or droplets of solute is produced by controlled rapid heating of the capillary tube connecting the HPLC column to the mass spectrometer. The solute droplets travel rapidly through the ion source, where they continue to vaporize and are ionized by standard chemical-ionization techniques. Modifications for fast atom bombardment MS and an atmospheric pressure ionization technique are also available. Such equipment has been applied to some relatively involatile lipids, such as phospholipids and gangliosides. In general, direct-inlet interfacing methods are better suited to microbore or capillary column HPLC applications where the volume of mobile phase is relatively limited.
With HPLC-MS systems, most structural information is obtained with lipids of comparatively low molecular weight; with compounds of lower volatility, it is sometimes possible to obtain molecular weights only (although this information is not negligible). At this time, the equipment is still very costly, but such is the pace of technological advance that this may not always be so. The use of systems of this kind in the analysis of lipids, especially for the identification of molecular species within specific lipid classes, is certainly becoming much more widespread [14,60]. Among recent applications, a moving-belt interface was used in conjunction with chemical ionization in analyses of neutral glycosphingolipids [27], HPLC-fast atom bombardment-MS interfaces were used to study acylcarnitines [94], coenzyme A esters [93] and glycosphingolipids [124,125], thermospray-MS interfaces have been utilized in analyses of phospholipids [1,53,54,56,96], diradylglycerol derivatives prepared from phospholipids [10,63], eicosanoids [1,55,56], fatty acid hydroperoxides [55] and acylcarnitines [76,77], and HPLC linked to atmospheric pressure-ionization MS has been employed for serum cholesterol [129], fatty acid anilides [61] and glycolipids [62].
A recent paper in which an HPLC micro-column (100 mm × 0.3 mm i.d.) of silica gel was used for the separation of glycosphingolipids with analysis by mass spectrometry may indicate an important new direction for the technique [126]. Flow rates as low as 6 microlitres/min were possible and components amounting to only 160 ng were separated and identified.
The above discussion no longer represents the current state of the art. The author's book (written jointly with Xianlin Han) Lipid Analysis (4th edition) (Oily Press, Bridgwater) brings mass spectrometric detection up to date.
2. Radioactivity detectors
Methods for detecting radioisotopes are often much more sensitive than many spectrophotometric and other physical procedures, and the use of lipids labelled with the beta-emitters 14C, 3H and 32P has revolutionized the study of lipid biochemistry. Indeed, the ease and accuracy of quantifying radioactive lipids are such that these are frequently used in the development of new methods to test recoveries. It can even be advantageous to convert lipids to isotopically labelled derivatives for quantification purposes. It is of course possible to use discontinuous methods of liquid-scintillation counting for estimating radioactivity in fractions collected from HPLC columns, but continuous methods are more relevant to this review. Methods for direct monitoring of radioactivity in HPLC mobile phases can be classified as either homogeneous or heterogeneous counting systems. In the latter, the eluent is passed through a flow cell, packed with a solid scintillator such as yttrium silicate or cerium-activated lithium glass, and positioned in a scintillation counter. An advantage is that solutes are easily recovered for analysis by other methods. In homogeneous counting, the eluent from the HPLC column is mixed with a liquid scintillant before passing into the counter. One difficulty is that there are limitations on the range of solvents that can be incorporated into mobile phases with this technique; chloroform especially will cause strong quenching. The major disadvantage, however, is that the residence time of a sample in the flow cell must be short to maximize resolution, and this greatly reduces the number of counts that can register.
Depending on the mode of HPLC and the type of counting procedure, the minimum limits of detection can be as high as 800 dpm and 27,000 dpm for 14C and 3H, respectively. Better results can be obtained with the higher-energy emitter 32P, as in separations of labelled phospholipids. Conventional liquid-scintillation counters equipped with flow cells can be used as HPLC detectors, but purpose-built counters/detectors are preferable in that they are designed to eliminate or at least minimize chemiluminescence effects due to flow phenomena.
Because of the sensitivity and specificity of radioactivity detection, preparation of isotopically labelled derivatives is sometimes undertaken as an aid to quantification of specific lipids [14]. In a recent application, glycosphingolipids were oxidized with galactose oxidase and reduced with 3H-labelled sodium borohydride for HPLC separation and detection with a flow-through scintillation counter [88]. Others have used the technique for analyses of acyl-coenzyme A esters [140], molecular species of phospholipids after conversion to 32P-labelled dimethylphosphoric acid esters [52] and molecular species of cholesterol esters [130]. Reviews of radiotracer techniques and HPLC in the analysis of fatty acids and eicosenoids [5] and phospholipids [83] have appeared.
3. Density, electrochemical and other detectors
A detector in which the changes in the density of a mobile phase on passage of a solute is sensed by variations in the frequency of a quartz oscillator has been applied in the analysis of simple lipid classes [131]. It appears to lack sensitivity and shares many of the disadvantages of differential refractometry.
An electrochemical (tensammetry) detector for phospholipids has been described, which has novelty value but apparently little else to commend it [6]. Much more valuable is the use of electrochemical detection with a mercury drop or glassy carbon electrode for the detection of lipid hydroperoxides [30,72,143]. The detector is sensitive and highly specific, since it is dependent on the controlled reduction of hydroperoxides to the analogous hydroxy compounds and can be used to quantify the former even when the two types of compound have co-eluted as is often the case. With the glassy electrode, the detection limit was ten times better than UV detection of the conjugated double bond systems [72].
An electron-spin resonance spectrometer has been used as a detector for an HPLC system to study the formation of free radicals from polyunsaturated fatty acids [123].
Transport-flame Ionization Detectors
1. Apparatus
A transport-flame ionization detector was first introduced by Pye Unicam Ltd. and, as with the evaporative light-scattering detector (Section E below), it is of universal applicability. In this commercial model, the eluent was fed continuously onto a clean moving wire that was passed into an evaporator oven to remove the solvent and then into a flame ionization chamber, similar to that in a gas chromatograph, where the solutes were combusted and detected. Subsequently, a more sensitive model was constructed in which the solute was pyrolysed to carbon dioxide and reduced to methane before entering the detector. These instruments were soon discontinued, apparently because they were unreliable and were not sufficiently sensitive, although second-hand equipment was in demand in some quarters for many years.
Many other detectors of this type have been built in laboratories but never manufactured commercially. For example, Privett and colleagues constructed and described a detector of improved design of this type, in which the eluent was entrained on a helical wire, so that more of the sample was combusted and the sensitivity of detection was greatly increased [122]. They later replaced the helical wire with a stainless-steel belt of perforated structure that allowed most or all of the eluent to be trapped and carried into the detector [103]. In addition, after evaporation of the mobile phase, the solute was converted to hydrocarbons in a stainless-steel reactor before being combusted and detected in a hydrogen flame. It has been shown to have a good linear response with respect to sample size although it was not as sensitive as some alternatives. The author was favourably impressed on seeing it in operation in 1985. Further development was been undertaken by a Danish manufacturer, who appear to have encountered some technical difficulties such that a commercial version was never released.
The only commercial detector based on this principle, for which there is a body of users, is manufactured by Tracor instruments (Austin, Texas, U.S.A; UK distributor - Kemtronix Ltd, Compton, Berks.), and details of its construction and some applications have been published [26]. A key component is a fibrous quartz belt around the circumference of a rotating disc, and enclosed in a heated ventilated housing, containing the solvent applicator and a dual-flame ionization detector. In operation, the mobile phase from the HPLC column is applied to the rotating belt and the volatile solvents are vaporized and eliminated by a vacuum pump, before the involatile solutes are transported into the twin flames of the detector, where they are combusted and ionized. As in a gas chromatograph, the total ion current is amplified and transmitted to a recorder and integrator. Any residual solute is burnt off in a pyrolyser unit before the belt picks up fresh eluent. While this detector is costlier than many others, this would not be a serious disadvantage if reliable results were possible. Regretfully, there were practical problems (see below), and this detector was also discontinued.
An interesting detector of the transport-flame ionization type has been described in which the transport mechanism was in effect a spoked wheel; the eluent from the HPLC column was deposited in discrete packets on the spokes, which were made from a material of low conductivity and thermal mass (quartz rods) and allowed the solvent to be evaporated in an air-flow of controlled temperature [71]. Eventually each rod was passed through the centre of the flame of the detector and ions were generated.
Most investigators, though working with a range of models, have reported that the response of the transport-flame ionization detector is linearly related to the amount of solute, although some correction factors might have to be introduced to compensate for non-combustible moieties in lipids. The only limitations on the choice of solvent for mobile phases are that they must be reasonably volatile and should not contain inorganic ions. Although the solute is destroyed by the detector, a stream-splitter can be introduced to divert a substantial proportion of the eluent to a collection system.
The author once gave his opinion that "if a reliable and relatively inexpensive detector of this type were to be made available commercially, it would fulfil a long-felt need. Indeed, it would not be an overstatement to suggest that it would revolutionize the practice of lipid analysis" [14]. This is probably true still, but there may yet be a long wait for such a detector. In my opinion, evaporative light-scattering detectors have reached a higher state of development and are a better investment.
2. Applications of transport-flame ionization detectors
The early literature on the subject of applications of transport-flame ionization detectors has been reviewed elsewhere [14] and is of limited relevance, since most workers used instruments that are no longer available commercially. Therefore, only applications of the Tracor™ detector need be considered here (although this too is no longer available commercially, it is still in use in some labs). Hammond [36] has briefly described his experience of this instrument in different forms of lipid analysis. In particular, he stresses the importance of setting up the applicator jet and the flame in the pyrolyser oven to the optimum values in order to realize the full potential of the instrument. Difficulties were experienced with lipids of relatively low molecular weight, which tended to evaporate before detection. Ions of sodium or potassium in solvents or samples were particularly troublesome, because of ionization of the flame and irreversible contamination of the belt. Perhaps surprisingly, silver ions eluting onto the belt did not appear to cause difficulties. A need for careful distillation of even "HPLC-grade" solvents in order to eliminate involatile impurities, as a prerequisite for acceptable base-line stability, has been reported by others [95]. In more recent instruments, the manufacturers appear to have improved background noise levels by improved electronic filtering.
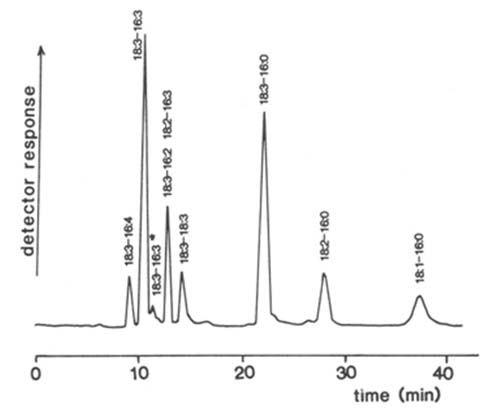
The first published application of the Tracor™ detector to lipids was for the separation of molecular species of phosphatidylglycerol and galactosyldiacylglycerols from plant chloroplasts by means of HPLC in the reversed-phase mode [91,95,112]. For the former the mobile phase, methanol-acetonitrile-ethylpropylamine-acetic acid (34.7:64.5:0.3:0.5 by volume), contained ionic species but gave a stable base-line. An application to molecular species of digalactosyldiacylglycerols is illustrated in Figure 2. Minor constituents, i.e. those present at a level of as low as 1.2 nmol, were quantifiable. Although the linearity of the detector response was not determined rigorously, direct quantification by integration of the signal gave results which were comparable to those obtained by alternative methods, and there did not appear to be any requirement for calibration factors to compensate for differences in chain length or degree of unsaturation. Similar methods were utilized to separate molecular species of the relatively uncommon diacylglyceryltrimethylhomoserine of algae [92].
Figure 2. Separation of molecular species of digalactosyldiacylglycerols from Dunaliella salina by HPLC in the reversed-phase mode with flame-ionization detection [112]. A column of Rainin Microsorb™ was employed with methanol-water (24:1, v/v) at a flow-rate of 0.8 mL/min as the mobile phase. (Reproduced by kind permission of the authors and of the Journal of Chromatography, and redrawn from the original paper).
Molecular species of triacylglycerols from milk fat have also been resolved by HPLC in the reversed-phase mode with flame-ionization detection with the Tracor instrument [95]. Once a number of practical difficulties associated with the detector were resolved, the response was found to be linear over a wide range of solute levels above 5 micrograms. Below this value, there was some drop in response thought to be associated with evaporation of the sample prior to combustion. In addition, the response was dependent on the nature of the fatty acid constituents in the molecular species. Similar results were described for separations of molecular species of triacylglycerols separated by silver ion chromatography [36]. While substantial differences in response according to degree of unsaturation were noted in this study, no such problems were described in a more extensive description of the technique [47]. The response of the Tracor detector was linear over a wide range and it was possible to use the instrument for routine analyses of confectionery fats in an industrial context.
A further important area for HPLC is the separation of lipid classes. Maxwell et al. [73] appear to have been the first to describe an application of the Tracor™ detector to this problem. They used a column of silica gel and a complex gradient system of dichloromethane-hexane to dichloromethane-chloroform followed by the introduction of ammonia in order to separate lipids ranging in polarity from sterol esters to phosphatidylcholine. The repeatability of injections was found to be good, and the detector response was linear for each component over a wide range of practical concentrations. Some variation in response that depended on the nature of the lipid classes was observed. Others [33] used a related system but with a gradient of chloroform to chloroform-methanol-ammonia to separate triacylglycerols and the individual phospholipids of soybean lecithin. Moreau and colleagues [84,85] adapted a method described earlier by Christie [12] for evaporative light-scattering detectors (discussed in greater detail in Section E.2 below) to the separation of plant lipids with flame-ionization detection. With a column of silica gel and a complex ternary gradient system, individual simple lipid classes were eluted first followed by glycolipids and then by each of the phospholipids. Although some base-line drift was evident, the progress of the separation was clearly monitored (see also Figure 7 below). The response of the detector was close to linear over a range of 1 to 200 micrograms for most lipid classes, but this did not appear to be true for triacylglycerols or free fatty acids. Some loss through evaporation of the last appears inevitable. Hammond has reported some success with similar methodology without giving full details [36].
The Tracor™ detector was used to quantify monomer, dimer and polymeric acids in commercial "dimer" mixtures following HPLC separation on a column of silica gel [137]. With careful calibration, it was possible to obtain results of good reproducibility, but the response factors for each component were very different. That for the dimer was 2.35 times greater than the factor for the monomer, for example. On the other hand, linear responses and small differences only with structural features were observed for cholesterol and its oxidation products, separated by HPLC on silica gel and with flame-ionization detection [70].
It is easy to exaggerate the problems of using the Tracor™ and related detectors for quantification of lipids, especially those associated with the variation in response due to small structural differences in the nature of the fatty acyl constituents. For example, spectrophotometric detectors have very little to offer in comparison, other than ready availability in laboratories [85]. After calibration with suitable standards and careful purification of mobile phases, transport flame-ionization detectors are being used for routine quantification in analyses of various kinds. It is also possible to insert a stream splitter between the end of the column and the detector to collect fractions for analysis and quantification by other means, and this is a satisfactory approach in many research applications especially.
Evaporative Light-Scattering Detectorsh5>
1. Construction and the nature of the response
An optical detection system for HPLC that is now assisting lipid analysts to make important advances has been variously termed a "mass detector", "evaporative analyser" or "light-scattering detector". The term "evaporative light-scattering detector" seems most accurate if rather lengthy; the pithy expression "mass detector" is easily confused with mass spectrometry by the unwary. With the instrument first described by Charlesworth [11] in 1978, the solvent emerging from the end of the column is evaporated in a stream of air or nitrogen in a heating chamber (Fig. 3); the solute does not evaporate, but is nebulized and passed in the form of minute droplets through a light beam, which is reflected and refracted. The scattered light is measured by a photomultiplier tube, set at an appropriate angle, and bears a relationship to the amount of material in the eluent. Such detectors are truly universal in their applicability, in that they will respond to any solute that does not evaporate before passing through the light beam.
Figure 3. A schematic diagram of an evaporative light-scattering detector.
The first commercial detector based on this principle is very similar in construction to the illustration and is available at a cost comparable to that of other optical detectors from Applied Chromatography Systems (ACS) Ltd. (Macclesfield, Cheshire, U.K.). There are at least three competitors now including instruments from Varex Corp. (Burtonsville, MD, U.S.A.), Cunow S.A. (Cergy St. Cristophe, France) and Sedere (Vitry-sur-Seine, France), some of which have distinctive design features with the reported objectives of improving sensitivity and linearity. For example in the Cunow detectors, the larger droplets in the spray from the nebulizer are condensed out before they reach the heater chamber; the consequence is a more uniform particle size and improved linearity. The Varex detector has a laser light source and a photodiode detector instead of a photomultiplier tube. Real differences therefore exist among commercial instruments and no objective comparison has yet been made. There are no special wavelength requirements for the light source, and in the first commercial instruments, it was a simply a lamp for a slide projector. On the other hand, a prototype experimental detector has been described that uses a laser light source [118,120], and at least one commercial instrument has this feature.
Evaporative light-scattering detectors can be considered to be universal in their applicability, in that they will respond to any solute that does not evaporate before passing through the light beam. Almost any solvent, including ketones, esters and chlorinated and aromatic compounds, can be used in complex gradients; up to 20% of water and small amounts of ionic species are also permissible. In designing an elution system to effect a particular separation, the analyst can therefore make use of the complete range of solvent selectivity groups as defined by Snyder [113,114] and recently re-evaluated [107]. Further advantages are that such detectors are simple, versatile and rugged in use, requiring little time for the base-line to stabilize when started up at the beginning of the day or when columns and mobile phases are changed; little or no adjustment may be required during lengthy periods of operation even with complex gradients. The sensitivity in the newer instruments is at least equal to that of the very best refractive index detectors, and is better than in transport-flame ionization detectors or in UV spectrophotometric detectors operated at low wavelengths. A further advantage is that evaporative light-scattering detectors are not affected by changes in ambient temperature or small variations in the flow-rate of the mobile phase.
As with all detectors, there are some disadvantages. Although the detector is destructive in that the sample is lost in the stream of air, it is possible to insert a stream splitter between the end of the column and the detector to divert much of the eluent to a collection device. A source of dry, filtered compressed air, that is capable of delivering 5 litres/min, is required and in practice, this means that an air compressor must be used; a standard cylinder of air or nitrogen is emptied in about 4 hours at this rate. For safety reasons (and to avoid the risk of litigation), most manufacturers recommend the use of nitrogen or carbon dioxide as evaporator gas when inflammable solvents are employed, but this is impracticable for prolonged usage. In practice, the concentration of organic solvent in the gas stream at any given moment should be very low and the risk of explosion with air as the carrier gas is minimal. The stream of air containing the evaporated solvent must be conducted directly to the outside of the laboratory or into a fume cupboard.
There are few limitations on the range of solvents that are suitable, and in particular they must be sufficiently volatile to evaporate in the heating chamber; the author [12] has used isopropanol containing 15% water without difficulty, but formic or acetic acids at a level of about 1% caused "spikes" on the base-line. Small but significant amounts of salts or ionic materials can be incorporated as ion suppressants into the mobile phases, without adversely affecting base-line stability, when the detector is operated at optimum sensitivity levels [13]. Indeed, there has even been a report of the use of 0.1M aqueous ammonium acetate as a mobile phase, though at the cost of a tenfold reduction in detector sensitivity [86]. Limited experience in the author's laboratory with the Cunow detector indicates that the aqueous component and any ionic species condense out preferentially in advance of the evaporator chamber, with the result that the base-line remains stable even at high sensitivity settings.
A key factor with all detectors is their linearity with respect to sample size, and investigators working with different evaporative light-scattering detectors have described the response variously as linear, sigmoidal or exponential. In the most comprehensive practical and theoretical investigations, use was made of the ACS detector but the results appear to be applicable to other commercial systems [90,97], and they compared well with data obtained from a custom-built detector with a laser as the light source [118,120]. It was observed that the detector response increased sigmoidally with increasing sample concentration, in a manner that could be predicted by changes in the size distribution of particles in the aerosol. Thus at low solute concentrations, the solute particles scattered light to a proportionately lesser extent. As the diameter of the droplets began to approach the wavelength of light, they no longer affected its passage and the response fell off rapidly. The detector response was close to linear over a concentration range of about two decades before tending to plateau.
To maximize the response and the linear range, it was necessary to adjust the flow-rate of the nebulizer gas and the temperature of the evaporator chamber to the optimum to give aerosol particles that were relatively uniform in size. However, small variations from the optimum temperature were found to have comparatively little effect on the response, which was relatively constant at fixed gas pressures. The same response was obtained for a given solute when the mobile phase was toluene, 2-butanone or tetrahydrofuran. In general, it was concluded that solvent viscosity was of relatively little importance, although anomalous results were obtained with dichloromethane, apparently because of its much greater density. Finally, the response was dependent on the refractive index of the sample and thus might be expected to vary with changes in structural features of lipids. Different lipid classes therefore require different response factors, although it is the author's experience (and that of others [119]) that the chain length and degree of unsaturation of the acyl constituents do not have a significant effect on response.
In summary, good quantitative results could be obtained if the instrumental parameters were adjusted carefully, and if the calibration conditions were rigidly set to be the same as in the analysis of real samples. An important note for manufacturers was that the design of the nebulizer was crucial, and this should be constructed to produce particles of uniform size in the optimum range. For lipid analysts, the eventual test is how these conclusions are borne out in practice with real samples, and happily they have been confirmed by such empirical studies (see below). In the custom-built detector incorporating a laser light source, the response was found to be related to the solute mass raised to the power of 1.35 [99,118,120]. This relationship has proved useful in a number of analytical circumstances and at least one manufacturer is offering a "linearizer" in which this formula is used electronically to "improve" the results.
2. Applications of light-scattering detection in lipid class separations
One of the more important problems in lipid analysis is the separation of the classes of tissue lipids, which at the extremes of polarity consist of cholesterol esters (hydrocarbon-like) and lysophospholipids (water-soluble) with a broad spectrum of simple and complex lipids with differing properties in between. Considerable progress was made with the aid of the evaporative light-scattering detector in the author's laboratory [12]. The initial objective was to separate and quantify the more abundant lipid classes in animal tissues, on the 0.2 to 0.4 mg scale and in as short a time as could conveniently be managed. The ACS mass detector was utilized, together with a ternary solvent delivery system and a short (5 × 100 mm) column packed with Spherisorb™ silica gel (3 micron particles). When solvents were selected for the mobile phase, the choice was constrained by the need for sufficient volatility for evaporation in the detector under conditions that did not cause evaporation of the solute, and at first by a perceived necessity to avoid ionic species, which would not evaporate. Similar restrictions would have applied with detectors operating on the transport-flame ionization principle.
To achieve the desired separation, it was necessary to use a complicated ternary-gradient elution scheme with eight programmed steps, starting with isooctane to separate the lipids of low polarity and ending with a solvent containing water to elute the phospholipids; a solvent of medium polarity was then needed to mediate the transfer from one extreme to the other, and mixtures based on isopropanol gave satisfactory results. The three solvent mixtures selected by trial and error were isooctane-tetrahydrofuran (99:1, v/v)(A), isopropanol-chloroform (4:1, v/v)(B) and isopropanol-water (1:1, v/v)(C). In the first successful elution scheme, a gradient of B into A was created to separate each of the simple lipids, then a gradient of C into A plus B was produced to separate each of the complex lipids; finally, a gradient was generated in the reverse direction to remove most of the bound water and to re-equilibrate the column prior to the next analysis. A high flow-rate (2 mL/min) appeared to assist the separation greatly, perhaps compensating for the absence of strong acid or inorganic ions, which others have found necessary for the separation of phospholipids. However, it was eventually found to be advantageous to add small amounts of organic ionic species (see below) [13].
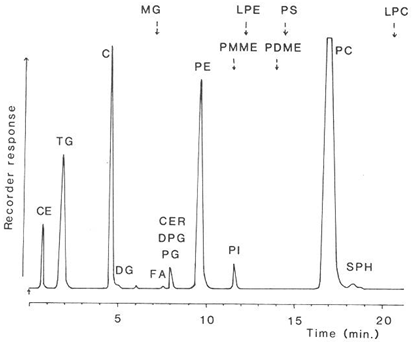
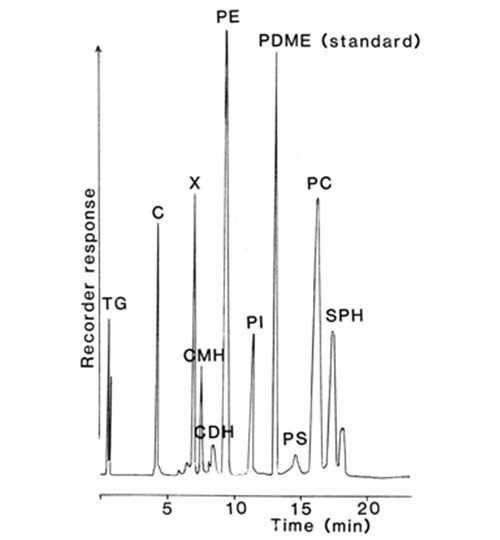
The nature of the separation achieved with the total lipids extracted from rat liver is shown in Figure 4. In spite of the abrupt changes in the composition of the mobile phase at various times, no disturbance of the base-line was apparent, and each of the main simple lipid and phospholipid classes was cleanly resolved in only 20 minutes. Only the highly acidic phospholipids, phosphatidic acid and to a lesser extent phosphatidylserine did not give satisfactory peaks. No "solvent peak" was apparent at the start of the analysis, as is often seen with other detectors, and di-tert-butyl-p-cresol (BHT) added to the extract as an antioxidant evaporated with the mobile phase and did not interfere. After a further 10 minutes of elution to remove bound water from the silica gel to restore its activity, the next sample could be analysed.
Figure 4. Separation of lipids (0.35 mg - injected in 5 microlitres of solvent) from rat liver on a column (100 × 5 mm) of silica gel (Spherisorb™ - 3 micron) with evaporative light-scattering detection (ACS model) [12]. (Reproduced by kind permission of the Journal of Lipid Research). The elution times of lipids not present in this particular sample are indicated. Abbreviations: CE, cholesterol esters; TG, triacylglycerols; C, cholesterol; DG, diacylglycerols; FA, free acids; CER, cerebrosides; PG, phosphatidylglycerol; DPG, diphosphatidylglycerol; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PC, phosphatidylcholine; SPH, sphingomyelin; MG, monoacylglycerols; LPE, lysophosphatidylethanolamine; PS, phosphatidylserine; LPC, lysophosphatidylcholine; PMME, phosphatidylmonomethylethanolamine; PDME, phosphatidyldimethylethanolamine.
It was subsequently shown that much better resolution of the minor acidic components could be obtained by adding small amounts of ionic species to the aqueous component of the mobile phase [13]. In addition, the lifetime of the column was greatly extended by this simple step. The optimum results were obtained in practice with 0.5 to 1 mM serine buffered to pH 7.5 with triethylamine. Perhaps surprisingly, ionic species at such concentrations had virtually no effect on the base-line of the detector, although the response changed somewhat and re-calibration was necessary. A further change was made to replace isooctane with hexane in the mobile phase, in order to reduce the maximum operating pressure required.
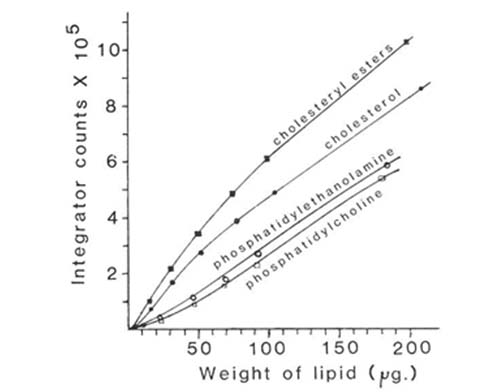
When evaporative light scattering detectors are used directly in quantitative analysis, it is necessary to work out the optimum conditions for the desired separations first and then carry out a calibration with lipid standards which are as close as possible in composition to the material to be analysed. The operating parameters for the instrument, such as gas pressure, evaporator temperature, and attenuation, must also be rigorously standardized. If the elution conditions or detector settings have later to be changed for any reason, a tedious re-calibration might be necessary. Figure 5 shows calibration curves for some of the main lipid classes in the analysis described. A different line was obtained for each lipid. With most, the response of the ACS mass detector was approximately rectilinear in the range 50 to 200 micrograms, but it tended to fall off rapidly below 10 micrograms. Rather better results have been obtained with the newer instruments on the market, both in terms of linearity and sensitivity (see below). However, with careful calibration, the data obtained in lipid analyses of this kind with the ACS detector were found to be at least as reliable in terms of accuracy and reproducibility as those from any other analytical method in use with lipids [12].
Figure 5. Calibration curves of detector response against amount of sample injected for some lipid classes separated by HPLC with evaporative light-scattering detection (ACS model) [12]. (Reproduced by kind permission of the Journal of Lipid Research).
It is also possible to use an internal standard to improve direct quantification with evaporative light-scattering detectors. For example, a synthetic phospholipid, phosphatidyldimethylethanolamine (dipalmitoyl), was used in this way in order that the absolute amount of phospholipid in an extract could be determined in addition to the relative compositions of each phospholipid class [23]. This lipid is only present naturally at very low levels in tissues so endogenous material does not interfere with the standard. The method was applied to determine the phospholipids of milk, which comprise only about 0.5% of the total lipids, and in order to analyse them effectively it was necessary to first obtain a concentrated fraction by a solid-phase extraction method before subjecting this to HPLC as shown in Figure 6. The chromatographic system employed the ACS mass detector and the ternary-gradient elution scheme described above.
Figure 6. HPLC separation of a phospholipid fraction from cow's milk on silica gel with a ternary elution scheme and evaporative light-scattering detection (ACS model); phosphatidyldimethylethanolamine was the internal standard [23]. (Reproduced by kind permission of the authors and of the Journal of the Society of Dairy Technology). See the legend to Figure 4 for a list of abbreviations (X is an unknown).
Recently others described a modified gradient elution procedure based on the above to improve the resolution of phospholipid components [67]. In this work, the Varex ELSD II light scattering detector, which is capable of much higher sensitivity, was employed but calibration curves were obtained that were similar in shape to those in Figure 5, if differing appreciably otherwise. N-Oleoylethanolamine was utilized as an internal standard. The original procedure has been adapted somewhat for the analysis of phospholipids alone, in this instance for detection with a Cunow LSD Model 10 light-scattering detector [48]. Cholesterol was employed as internal standard, and the response of the detector for each phospholipid relative to cholesterol was linear over a wide range. Similar findings had been reported earlier for a Cunow detector with a different gradient elution system in analyses of phospholipids [7].
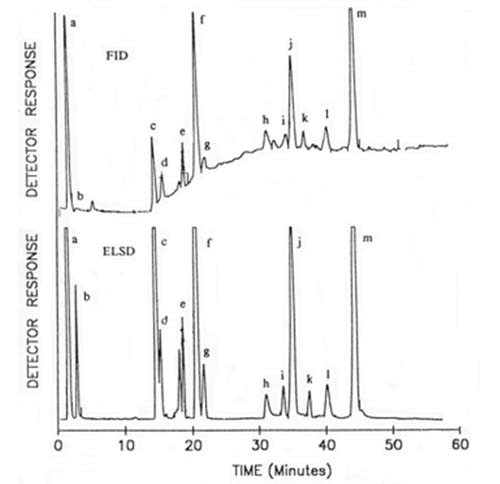
The author [22] modified his own method in order to better resolve the distinctive phospholipids and glycosyldiacylglycerols found in plant and especially cereal tissues. To elute sequentially mono- and digalactosyldiacylglycerols and N-acyl-phosphatidylethanolamine and their lyso forms, hexane-butan-2-one-acetic acid (35:65:0.4, v/v/v) was used as part of a gradient before the more usual phospholipids emerged with the elution scheme described above. Following an earlier adaptation of this methodology for the Tracor transport-flame ionization detector (see Section D.2), Moreau [84] demonstrated that much better results could be obtained with the Varex light-scattering detector and a comparison of the two is illustrated in Figure 7. The improved sensitivity and base-line stability of the latter are very evident. In this instance, no chloroform was required in the mobile phase (as there was no need to resolve sphingomyelin) and additional gradient steps were introduced to separate each of the glycolipids ahead of the phospholipids. Brief details only of a completely different elution scheme for the separation of both simple and complex lipids in wheat flour have been published; a quaternary gradient system was required, starting with toluene containing 0.125% formic acid, and proceeding via ethyl acetate, methanol and water-based mixtures [37]. In this instance, the ACS mass detector was utilized.
Figure 7. Comparison of the analysis of lipid classes from corn coleoptiles by HPLC with transport-flame ionization detection (FID) (Tracor model) and evaporative light-scattering detection (ELSD) (Varex model) (B), each with 125 micrograms of lipid in total [84]. Reproduced by kind permission of the author and of Portland Press. Abbreviations: a, sterol esters; b, triacylglycerols; c, sterols; d, free fatty acids; e, acylated sterol glycosides; f, monogalactosyldiacylglycerols; g, sterol glycosides; h, digalactosyldiacylglycerols; i, cardiolipin; j, phosphatidylethanolamine; k, phosphatidylglycerol; l, phosphatidylinositol; m, phosphatidylcholine.
Many lipid analysts have used evaporative light-scattering detection and columns of silica gel to separate phospholipid classes in the absence of simple lipids. For example, Stolyhwo et al. [121] used a gradient of ammonia in methanol in essence to obtain excellent resolution of phospholipid classes from plasma, and from rapeseed and soybean lecithins. Others used a similar elution system containing an appreciable concentration of chloroform [4]. In studies of both preparative [45,134,135] and analytical [25,136] scale separations of phospholipids, various gradients of hexane-isopropanol-water and increasing in polarity were generated for specific purposes.
Similarly, it has proved possible to use light-scattering detection and gradients of isopropanol in isooctane to separate all the simple lipid classes (and not the phospholipids) in some samples of commercial interest [8]. In other work, a column containing an octadecylsilyl-bonded phase was employed to separate simple lipid classes, i.e. tri-, di- and monoacylglycerols and free acids, with some simultaneous fractionation into molecular species; the mobile phase here was a gradient of acetonitrile, acetone and dichloromethane [25].
Much remains to be done to improve the conditions for the separation of lipid classes and especially for the simultaneous separation of simple and complex lipids by means of HPLC. Many more solvent combinations must be tried, and more column packing materials must be tested for the purpose. However, as evaporative light-scattering detectors appear in more laboratories, there is now a solid foundation upon which to build.
3. Applications of light-scattering detection in separations of molecular species of lipids
Because of their commercial importance, methods for the separation of molecular species of triacylglycerols have been studied with particular intensity, most analysts preferring to work with reversed-phase systems. ODS columns have been used universally with those with a high carbon content generally being preferred. While acetonitrile is an essential component of the mobile phase, it must contain a substantial proportion of a non-polar modifier to solubilize the lipid and displace it from the stationary phase. There is no consensus as to which is the best modifier solvent and the choice has often been determined by the availability of a particular detector. There is no such practical limitation with the evaporative light-scattering detector, which has been shown to be at least as sensitive as any other to have been used for the purpose, and exhibited no base-line drift, even with lengthy gradients [34,41,98-100,105,106,119,132,133]. When solvent restrictions are removed, it is also easier to ensure that there are no problems over the solubility of the solute in the mobile phase as has sometimes been observed [14].
In nearly all the published work to date, the Applied Chromatography Systems (ACS) detector has been used. As with the lipid class separations discussed above, Robinson and Macrae found that it was essential for all the instrumental and chromatography parameters, such as nebulizer inlet pressure, evaporator temperature, mobile phase and flow-rate, to be standardized prior to analysis, since changes in any of these could affect the response [105]. The detection limits were of the order of 1 microgram for most triacylglycerol standards, but much better results can now be obtained with the newer commercial instruments. As expected, the response with respect to mass was found to be sigmoidal for all triacylglycerol standards, but was approximately linear in the range 10 to 25 micrograms for saturated compounds (limited by solubility problems with the mobile phase selected for the work), and in the range 10 to 60 micrograms for unsaturated compounds. The logarithm of the detector response was rectilinear with respect to the logarithm of sample mass. However, the slopes of the lines for different standards were not found to be parallel, and there was no obvious relationship to fatty acyl structure.
This has not been the author's experience or that of others [41,119], who have found very little effect of the nature of the fatty acyl group on the response. For example, with the same type of detector, Herslof and Kindmark [41] obtained an identical detection limit, but the reproducibility of the analysis of molecular species of triacylglycerols from soybean oil was found to be quite acceptable, even with gradient elution; relative standard deviations of 2% were obtained for the main components. One reason for the better results obtained in the latter work was that the data were expressed in terms of the relative proportions (percentages) of the different components, and not in terms of the absolute amounts of each. Similar data were reported by others, but with greater variation for minor components [34]. Again better results would be anticipated with the newer commercial instruments.
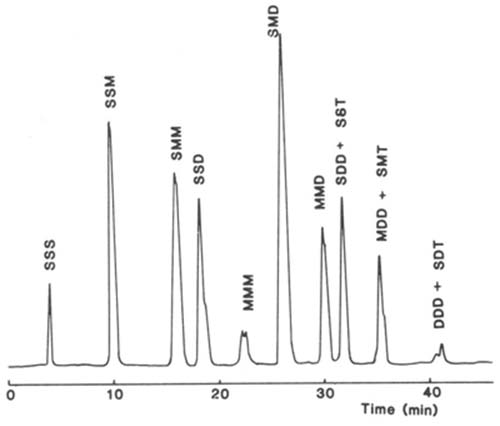
Silver ion chromatography of triacylglycerols with light-scattering detection has been employed to good effect in the author's laboratory. The stationary phase consisted of an ion-exchange medium, which was a silica gel matrix with bonded phenyl sulphonic acid moieties (Nucleosil™ 5SA), and it was converted to the silver ion form by injection of silver nitrate into an aqueous mobile phase via the Rheodyne™ valve [15] (discussed in greater detail on this website here..). With this column, it was possible to separate molecular species according to the single property of degree of unsaturation. As an example, a separation of the triacylglycerols of rat adipose tissue on a silver ion column is illustrated in Figure 8 [16]. The trisaturated fraction is eluted first, followed by disaturated-monoenes, saturated-dimonoenes, disaturated-dienes, trimonoenes and so forth. With this system, one dienoic acyl group is retained by the equivalent of about 2.5 monoenes and one trienoic acyl group by the equivalent of about two dienes. Species containing γ-linolenic acid, as in evening primrose oil, were eluted before those with alpha-linolenic acid [18]. The mobile phase was a gradient of dichloromethane-dichloroethane (1:1, v/v) to acetone to elute the more saturated fractions, before acetonitrile was introduced to bring off polyunsaturated species. Of course, the light-scattering detector is ideal for such a combination of solvents.
By increasing the acetonitrile content, it was possible to obtain a comprehensive separation of triacylglycerols as unsaturated as that from linseed oil, where the predominant molecular species had nine double bonds, while species with up to 15 double bonds were separated from fish oils [16,65]. More subtle separations have also been reported. Thus with meadowfoam oil, which contains a number of different monoenoic fatty acids, species were resolved in which the only difference was the position of one double bond in one of the three fatty acids in the molecule [89].
The technique has been applied similarly to the separation of molecular species of cholesterol esters with light-scattering detection [44].
Figure 8. Separation of molecular species of triacylglycerols from rat parametrial adipose tissue on a column of Nucleosil™ 5SA (250 × 4.6 mm i.d.) in the silver ion form with light-scattering detection (ACS Model) [16]. The mobile phase consisted of a gradient of acetone into dichloroethane-dichloromethane (1:1, v/v) to elute the more saturated fractions before acetonitrile was introduced to bring off the polyunsaturated components. Abbreviations: S, saturated; M, monoenoic; D, dienoic; T, trienoic acyl groups. Reproduced by kind permission of the Journal of Chromatography.
The major virtue of silver ion chromatography in comparison to reversed-phase partition chromatography is that separation with the former is based on the single property of degree of unsaturation and fractions are intuitively easy to identify. With reversed-phase chromatography, in contrast, separation is according to both the combined chain lengths and the total number of double bonds in the fatty acyl groups, each double bond reducing the retention time by the equivalent of about two carbon atoms. Excellent resolution is possible, but it is not easy to identify fractions when unknown samples are analysed.When the two techniques are used to complement each other, analysts have a particularly powerful separatory tool. Silver ion chromatography provides separation by degree of unsaturation, and fractions collected can then be resolved by reversed-phase HPLC when resolution is then simply according to the combined chain lengths of the fatty acyl residues. The virtues of this approach, for HPLC with light-scattering detection, have been demonstrated for meadowfoam oil [89] and for some fish oils [64].
As yet the evaporative-light scattering detector has been little used for molecular species of phospholipids, although the potential has been indicated in one paper [115], and further applications will no doubt follow.
4. Some miscellaneous separations
HPLC with evaporative light-scattering detection has not been used for the analysis of fatty acids per se, as they are too volatile, but it has proved its worth as a micro-preparative tool for the isolation of fractions for analysis by other means. In such circumstances, a stream-splitter must be placed between the end of the column and the detector. For example, picolinyl ester derivatives of fatty acids were separated on the 1-2 mg scale by reversed-phase chromatography by gradient elution with mobile phases containing pyridine and acetic acid amongst other components [24]; fractions were subsequently analysed by GC-MS. It is not easy to see how any other type of HPLC detector could have coped with this solvent combination. Silver ion HPLC has been applied in a similar manner to the separation of methyl esters of fatty acids with zero to six double bonds, again as an aid to identification by GC-MS [2,19,20,116].
The light-scattering detector was utilized in the detection and quantification of tocopherols and phytosterols from seed oils separated by reversed-phase HPLC [139]. The response to each component differed somewhat but was essentially linear over the range 10 to 100 micrograms with an error of 2% over five analyses.
Gel permeation chromatography with light-scattering detection has been employed to separate and quantify polymers in oxidized fish oils [9]. In this instance, glycerol was used as an internal standard and careful calibration was necessary; although the response to the standard was almost linear over a wide range, this was not true of the polymer fraction.
Abbreviations: GC, gas chromatography; HPLC, high-performance liquid chromatography; IR, infrared; MS, mass spectrometry; ODS, octadecylsilyl; RI, refractive index; UV, ultraviolet.
Acknowledgement: This work has been funded in part by the Scottish Executive Environmental and Rural Affairs Department.
- Abian, J. and Gelpi, E., J. Chromatogr., 394, 147-153 (1987).
- Adkisson, H.D., Risener, F.S., Zarrinkar, P.P., Walla, M.D., Christie, W.W. and Wuthier, R.E., FASEB J., 5, 344-353 (1991).
- Aitzetmuller, K., J. High Resolut. Chromatogr., 13, 375-377 (1990).
- Becart, J., Chevalier, C. and Biesse, J.-P., J. High Resolut. Chromatogr., 13, 126-129 (1990).
- Birkle, D.L., Bazan, H.E.P. and Bazan, N.G., Prog. HPLC, 3, 11-26 (1988).
- Boswart, J., Schmidt, T., Kostiuk, P., Pacakova, V. and Stulik, K., J. Chromatogr., 495, 61-70 (1989).
- Breton, L., Serkiz, B., Volland, J.-P. and Lepagnol, J., J. Chromatogr., 497, 243-249 (1989).
- Bruns, A., Fat Sci. Technol., 90, 289-291 (1990).
- Burkow, I.C. and Henderson, R.J., Lipids, 26, 227-231 (1991).
- Butikofer, P., Kuypers, F.A., Shackleton, C., Brodbeck, U. and Stieger, S., J. Biol. Chem., 265, 18983-18987 (1990).
- Charlesworth, J.M., Anal. Biochem., 50, 1414-1420 (1978).
- Christie, W.W., J. Lipid Res., 26, 507-512 (1985).
- Christie, W.W., J. Chromatogr., 361, 396-399 (1986).
- Christie, W.W., High-Performance Liquid Chromatography and Lipids, Pergamon Press, Oxford (1987).
- Christie, W.W., J. High Res. Chromatogr. Chromatogr. Commun., 10, 148-150 (1987).
- Christie, W.W., J. Chromatogr., 454, 273-284 (1988).
- Christie, W.W., Gas Chromatography and Lipids, The Oily Press, Ayr (1989)
- Christie, W.W., Fat. Sci. Technol., 93, 65-66 (1991).
- Christie, W.W., Brechany, E.B. and Shukla, V.K.S., Lipids, 24, 116-120 (1989).
- Christie, W.W., Brechany, E.B. and Stefanov, K., Chem. Phys. Lipids, 46, 127-135 (1988).
- Christie, W.W. and Breckenridge, G.H.M., J. Chromatogr., 469, 261-269 (1989).
- Christie, W.W. and Morrison, W.R., J. Chromatogr., 436, 510-513 (1988).
- Christie, W.W., Noble, R.C. and Davies, G., J. Soc. Dairy Technol., 40, 10-12 (1987).
- Christie, W.W. and Stefanov, K., J. Chromatogr., 392, 259-265 (1987).
- Denoyer,C. and Girard, J.P., Ind. Aliment. Agric., 105, 515-517 (1988).
- Dixon, J.B., Chimia, 38, 82-86 (1984).
- Evans,J .E. and McCluer, R.H., Biomed. Envir. Mass Spectrom., 14, 149-153 (1987).
- Frede, E., Chromatographia, 21, 29-36 (1986).
- Frede, E. and Thiele, H., J. Am. Oil Chem. Soc., 64, 521-528 (1987).
- Funk, M.O., Free Radical Biol. Med., 3, 319-321 (1987).
- Gilkison, I.S., Chromatographia, 26, 181-185 (1988).
- Greenspan, M.D., Lo, C.-Y.L., Hanf, D.P. and Yudkovitz, J.B., J. Lipid Res., 29, 971-976 (1988).
- Grieser, M.D. and Geske, J.N., J. Am. Oil Chem. Soc., 66, 1484-1487 (1989).
- Grossberger, T. and Rothschild, E., LC-GC International, 2, 44-48 (1989).
- Hamilton, R.J., Mitchell, S.F. and Sewell, P.A., J. Chromatogr., 395, 33-46 (1987).
- Hammond, E.W., Chromatography and Analysis, October Issue, 7-9 (1988).
- Hammond, E.W., Trends Anal. Chem., 8, 308-313 (1989).
- Hanis, T., Smrz, M., Klir, P., Macek, K., Klima, J., Base, J. and Deyl, Z., J. Chromatogr., 452, 443-457 (1988).
- Heinze, T., Kynast, G., Dudenhausen, J.W., Schmitz, C. and Saling, E., Chromatographia, 25, 497-503 (1988).
- Herslof, B., J. High Resolut. Chromatogr., Chromatogr. Commun., 4, 471-473 (1981).
- Herslof, B. and Kindmark, G., Lipids, 20, 783-790 (1985).
- Holte, L.L., van Kuijk, F.J.G.M. and Dratz, E.A., Anal. Biochem., 188, 136-141 (1990).
- Homan, R. and Pownall, H.J., Anal. Biochem., 178, 166-171 (1989).
- Hoving, E.B., Muskiet, F.A.J. and Christie, W.W., J. Chromatogr., 565, 103-110 (1991).
- Huys, M., Van der Meeren, P., Vanderdeelen, J. and Baert, L., Med. Fac. Landbouww. Rijksuniv. Gent, 53, 1667-1677 (1988).
- Iwaoka, T., Tabata, F. and Takahashi, T., Free Radical Biol. Med., 3, 329-333 (1987).
- Jeffrey, B.S.J., J. Am. Oil Chem. Soc., 68, 289-293 (1991).
- Juaneda, P., Rocquelin, G. and Astorg, P.O., Lipids, 25, 756-759 (1990).
- Jungalwala, F.B., Evans, J.E. and McCluer, R.H., Biochem. J., 155, 55-60 (1976).
- Kaneko, T., Ohta, Y. and Machita, Y., Agric. Biol. Chem., 51, 2023-2024 (1987).
- Karger, B.L. and Vouros, P., J. Chromatogr., 323, 13-22 (1985).
- Kennerley, D.A., J. Biol. Chem., 262, 16305-16313 (1987).
- Kim, H.-Y. and Salem, N., Anal. Chem., 58, 9-14 (1986).
- Kim, H.-Y. and Salem, N., Anal. Chem., 59, 722-726 (1987).
- Kim, H.-Y. and Salem, N., Prostaglandins, 37, 105-119 (1989).
- Kim, H.-Y., Yergey, J.A. and Salem, N., J. Chromatogr., 394, 155-170 (1987).
- Kobayashi, T. and Goto, I., Biochim. Biophys. Acta, 1081, 159-166 (1991).
- Kondoh, Y. and Takano, S., J. Chromatogr., 393, 427-432 (1987).
- Kuksis, A. (Editor), Chromatography of Lipids in Biomedical Research and Clinical Diagnosis, Elsevier, Amsterdam (1987).
- Kuksis, A., Marai, L., Myher, J.J. and Pind, S., in Chromatography of Lipids in Biomedical Research and Clinical Diagnosis, pp. 403-440 (1987) (edited by A. Kuksis, Elsevier, Amsterdam).
- Kusaka, T., Ikeda, M., Nakano, H. and Numajiri, Y., J. Biochem. (Tokyo), 104, 495-497 (1988).
- Kushi, Y., Rokukawa, C., Numajir, Y., Kato, Y. and Handa, S., Anal. Biochem., 182, 405-410 (1989).
- Kuypers, F.A., Butikofer, P. and Shackleton, C.H.L., J. Chromatogr., 562, 191-206 (1991).
- Laakso, P. and Christie, W.W., J. Am. Oil Chem. Soc., 68, 213-223 (1991).
- Laakso, P., Christie, W.W. and Pettersen, J., Lipids, 25, 284-291 (1990).
- Lingeman, H., Underberg, W.J.M., Takade, A. and Hulshoff, A., J. Liq. Chromatogr., 8, 789-874 (1985).
- Lutzke, B.S. and Braughler, J.M., J. Lipid Res., 31, 2127-2130 (1990).
- McCluer, R.H., Ullman, M.D. and Jungalwala, F.B., Adv. Chromatogr., 25, 309-353 (1986).
- McCluer, R.H., Ullman, M.D. and Jungalwala, F.B., Methods Enzymol., 172, 538-575 (1989).
- Maerker, G., Nungesser, E.H. and Zulak, I.M., J. Agric. Food Chem., 36, 61-63 (1988).
- Malcolme-Lawes, D.J. and Moss, P., J. Chromatogr., 482, 53-64 (1989).
- Matsushita, S., Terao, J., Yamada, K. and Shibata, S.S., Basic Life Sci., 49, 163-174 (1988).
- Maxwell, R.J., Nungesser, E.H., Marmer, W.N. and Foglia, T.A., LC-GC, 5, 829-833 (1987).
- Mayr, G.W., Biochem. J., 254, 585-591 (1988).
- Mayr, G.W. in Methods in Inositol Research, pp. 83-108 (1990) (edited by R.F. Irvine, Raven Press, New York).
- Millington, D.S., Norwood, D.L., Kodo, N., Roe, C.R. and Inoue, F., Anal. Biochem., 180, 331-339 (1989).
- Millington, D.S., Roe, C.R. and Maltby, D.A., Biomed. Environ. Mass Spectrom., 14, 711-716 (1987).
- Minkler, P.E., Ingalls, S.T. and Hoppel, C.L., Anal. Biochem., 185, 29-35 (1990).
- Mita, H., Yasueda, H., Hayakawa, T. and Shida, T., Anal. Biochem., 180, 131-135 (1989).
- Miyazawa, T., Yasuda, K., Fujimoto, K. and Kaneda, T., Anal. Lett., 21, 1033-1044 (1988).
- Miyazawa, T., Yasuda, K., Fujimoto, K. and Kaneda, T., Basic Life Sci., 49, 191-194 (1988).
- Miyazawa, T., Fujimoto, K. and Oikawa, S., Biomed. Chromatogr., 4, 131-134 (1990).
- Morgan, R.O. and Catt, K.J., Prog. HPLC, 3, 27-55 (1988).
- Moreau, R.A., in Plant Lipid Biochemistry, Structure and Utilization, pp. 20-22 (1990) (edited by P.J. Quinn and J.L. Harwood, Portland Press, London).
- Moreau, R.A., Asmann, P.T. and Norman, H.A., Phytochemistry, 29, 2461-2466 (1990).
- Mourey, T.H. and Oppenheimer, L.E., Anal. Chem., 56, 2427-2434 (1984).
- Mullertz, A., Schmedes, A. and Holmer, G., Lipids, 25, 415-418 (1990).
- Myers, R.L., Ullman, M.D., Ventura, R.F. and Yates, A.J., Anal. Biochem., 192, 156-164 (1991).
- Nikolova-Damyanova, B., Christie, W.W. and Herslof, B., J. Am. Oil Chem. Soc., 67, 503-507 (1990).
- Norman, H.A. and St. John, J.B., J. Lipid Res., 27, 1104-1107 (1986).
- Norman, H.A. and Thompson, G.A., Arch. Biochem. Biophys., 242, 168-175 (1985).
- Norman, H.A. and Thompson, G.A., Plant Sci., 42, 83-87 (1985).
- Norwood, D.L., Bus, C.A. and Millington, D.S., J. Chromatogr., 92, 289-302 (1990).
- Norwood, D.L., Kodo, N. and Millington, D.S., Rapid Commun. Mass Spectrom., 2, 269-272 (1988).
- Nurmela, K.V.V. and Satama, L.T., J. Chromatogr., 435, 139-148 (1988).
- Odham, G., Valeur, A., Michelsen, P., Aronsson, E. and McDowall, M., J. Chromatogr., 434, 31-41 (1988).
- Oppenheimer, L.E. and Mourey, T.H., J. Chromatogr., 323, 297-304 (1985).
- Perrin, J.-L. and Prevot, A., Rev. Franç. Corps Gras, 33, 437-445 (1986).
- Perrin, J.-L., Prevot, A., Stolyhwo, A. and Guiochon, G., Rev. Franç. Corps Gras, 31, 495-510 (1984).
- Perrin, J.-L., Prevot, A., Traitler, H. and Bracco, U., Rev. Franç. Corps Gras, 34, 221-223 (1987).
- Postle, A.D., J. Chromatogr., 415, 241-251 (1987).
- Powell, W.S., in Prostaglandins and Related Substances; a Practical Approach, pp. 75-98 (1987) (edited by C. Benedetto, R.G. McDonald-Gibson, S. Nigam and T.F. Slater, IRL Press, Oxford).
- Privett, O.S. and Erdahl, W.L., Anal. Biochem., 84, 449-461 (1978).
- Rastegar, A., Pelletier, A., Duportail, G., Freysz, L. and Leray, C., J. Chromatogr., 518, 157-165 (1990).
- Robinson, J.L. and Macrae, R., J. Chromatogr., 303, 386-390 (1984).
- Robinson, J.L., Tsimidou, M. and Macrae, R., J. Chromatogr., 324, 35-52 (1985).
- Rutan, S.C., Carr, P.W., Cheong, W.J., Park, J.H. and Snyder, L.R., J. Chromatogr., 463, 21-37 (1989).
- Salari, H. and Eigendorf, G.K., J. Chromatogr., 527, 303-314 (1990).
- Shukla, V.K.S., Prog. Lipid Res., 27, 5-38 (1988).
- Shukla, V.K.S., J. Dispersion Sci. Technol., 10, 581-616 (1990).
- Shukla, V.K.S., Nielsen, W.S. and Batsberg, W., Fette Seifen Anstrichm., 85, 274-278 (1983).
- Smith, L.A., Norman, H.A., Cho, S.H. and Thompson, G.A., J. Chromatogr., 346, 292-299 (1985).
- Snyder, L.R., J. Chromatogr. Sci., 16, 223-234 (1978).
- Snyder, L.R. and Kirkland, J.J., Introduction to Modern Liquid Chromatography (2nd edition) (1979) (John Wiley & Sons, New York).
- Sotirhos, N., Thorngren, C. and Herslof, B., J. Chromatogr., 331, 313-320 (1985).
- Stefanov, K., Konaklieva, M., Brechany, E.Y. and Christie, W.W., Phytochemistry, 27, 3495-3497 (1988).
- Stokl, C. and Purdy, W.C., Anal. Lett., 21, 1-20 (1988).
- Stolyhwo, A., Colin, H. and Guiochon, G., J. Chromatogr., 265, 1-18 (1983).
- Stolyhwo, A., Colin, H. and Guiochon, G., Anal. Chem., 57, 1342-1354 (1985).
- Stolyhwo, A., Colin, H., Martin, M. and Guiochon, G., J. Chromatogr., 288, 253-275 (1984).
- Stolyhwo, A., Martin, M. and Guiochon, G., J. Liq. Chromatogr., 10, 1237-1253 (1987).
- Stolyhwo, A., Privett, O.S. and Erdahl, W.L., J. Chromatogr. Sci., 11, 263-267 (1973).
- Sugata, R., Iwahashi, H., Ishii, T. and Kido, R., J. Chromatogr., 487, 9-16 (1989).
- Suzuki, M., Sekine, M., Yamakawa, T. and Suzuki, A., J. Biochem. (Tokyo), 105, 829-833 (1989).
- Suzuki, M., Yamakawa, T. and Suzuki, A., J. Biochem. (Tokyo), 108, 92-98 (1990).
- Suzuki, M., Yamakawa, T. and Suzuki, A., J. Biochem. (Tokyo), 109, 503-506 (1991).
- Svensson ,L., Sisfontes, L., Nyborg, G. and Blomstrand, R., Lipids, 17, 50-59 (1982).
- Takano, S. and Kondoh, Y., J. Am. Oil Chem. Soc., 64, 1001-1003 (1987).
- Takatsu, A. and Nishi, S., Anal. Chem., 60, 2237-2239 (1988).
- Thomas, M.S. and Rudel, L.L., J. Lipid Res., 28, 572-581 (1987).
- Trathnigg, B. and Mittelbach, M., J. Liq. Chromatogr., 13, 95-105 (1990).
- Tsimidou, M. and Macrae, R., J. Chromatogr., 285, 178-181 (1984).
- Tsimidou, M. and Macrae, R., J. Chromatogr. Sci., 23, 155-160 (1985).
- Van der Meeren, P., Vanderdeelen, J., Huys, M. and Baert, L., J. Chromatogr., 447, 436-442 (1988).
- Van der Meeren, P., Vanderdeelen, J., Huys, M. and Baert, L., J. Am. Oil Chem. Soc., 67, 815-820 (1990).
- Van der Meeren, P., Vanderdeelen, J., Huys, M. and Baert, L., in Phospholipids, pp. 273-277 (1990) (edited by I. Hanin & G. Pepeu, Plenum Press, New York).
- Veasey, R.L., J. Am. Oil Chem. Soc., 63, 1043-1046 (1986).
- Vercaemst, R., Union, A., Rosseneu, M., De Craene, I., De Backer, G. and Kornitzer, M., Atherosclerosis, 78, 245-250 (1989).
- Warner, K. and Mounts, T.L., J. Am. Oil Chem. Soc., 67, 827-831 (1990).
- Watmough, N.J., Turnbull, D.M., Sherratt, H.S.A. and Bartlett, K., Biochem. J., 262, 261-269 (1989).
- Wood, R., Biochem. Arch., 2, 63-71 (1986).
- Yamada, K., Abe, S., Katayama, K. and Sato, T., J. Chromatogr., 424, 367-372 (1988).
- Yamada, K., Terao, J. and Matsushita, S., Lipids, 22, 125-128 (1987).
- Yamaguchi, M., Hara, S., Takemori, Y. and Nakamura, M., Anal. Sci., 5, 35-38 (1989).
- Yamamoto, Y. and Ames, B.N., Free Radical Biol. Med., 3, 359-361 (1987).
- Yamamoto, Y., Brodsky, M.H., Baker, J.C. and Ames, B.N., Anal. Biochem., 160, 7-13 (1987).
- Yamamoto,Y ., Frei, B. and Ames, B.N., Methods Enzymol., 186, 371-380 (1990).
In This Section
- Solid-phase extraction columns in the analysis of lipids
- Preparation of Ester Derivatives of Fatty Acids for Chromatographic Analysis
- Preparation of Lipid Extracts Tissues
- The Chromatographic Resolution of Chiral Lipids
- Detectors for HPLC of Lipids with Special Reference to Evaporative Lght-Scattering Detection
- Why Doesn't Your Method Work When I Try It?
- Laboratory Accreditation in a Lipid Analysis Context
- What Column do I Need for Gas Chromatographic Analysis of Fatty Acids?
- Fatty Acid Analysis by HPLC
- Alternatives to Methyl Esters for GC Analysis of Fatty Acids
- A Practical Guide to the Analysis of Conjugated Linoleic Acid (CLA)
- Application of Infrared Spectroscopy to the Rapid Determination of Total Saturated, trans, Monounsaturated, and Polyunsaturated Fatty Acids
- The Use of Lithiated Adducts for Structural Analysis of Acylglycerols by Mass Spectrometry with Electrospray Ionization
- Identification of FAME Double Bond Location by Covalent Adduct Chemical Ionization (CACI) Tandem Mass Spectrometry
- The Use of Countercurrent Chromatography (CCC) in Lipid Analysis
- Gas Chromatographic Analysis of Plant Sterols
- Analysis of Tocopherols and Tocotrienols by HPLC
- Reversed-Phase HPLC of Triacylglycerols
- Structural Analysis of Triacylglycerols
- Thin-Layer Chromatography of Lipids
- High-temperature Gas Chromatography of Triacylglycerols
- Modification of an AOCS Official Method for Crude Oil Content in Distillers Grains and Other Agricultural Materials
- Lipidomics - A Personal View