The Oxylipin Biosynthetic Pathways in Plants
The Authors: Florian Brodhun and Ivo Feussner
Oxylipins are oxidized fatty acids and metabolites derived therefrom. These lipids are abundant in mammals as well as in flowering plants, mosses, algae, bacteria and fungi [1]. They serve as signal molecules regulating developmental processes like pollen formation and mediate responses to the environment like wound healing and defensive reactions or have direct antimicrobial properties [2]. Oxylipin biosynthesis may may occur in plants at the level of esterified or free polyunsaturated fatty acids. For simplicity we will focus here on the first case. Then oxylipin biosynthesis starts with the release of fatty acids from membrane lipids by the action of a lipase. The next reaction is an initial oxidation reaction by different enzymes at different positions of the fatty acid backbone as indicated in Figure 1.
Figure 1. Overview of the oxylipin biosynthesis pathway in plants (abbreviations are explained in the text).
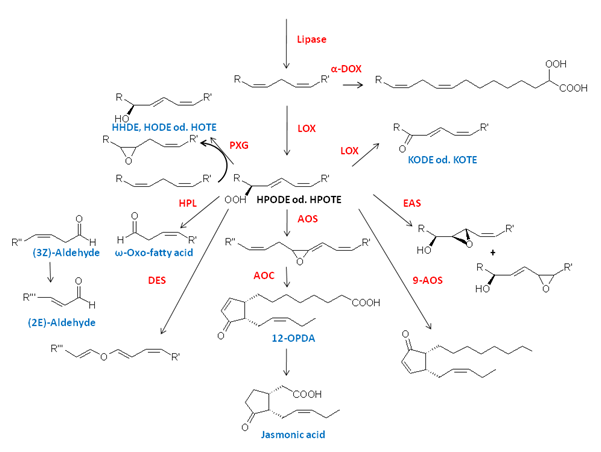
In the first step the free fatty acid is oxidized by molecular oxygen yielding a hydroperoxy fatty acid. This reaction can either be catalyzed by a lipoxygenase (LOX) or α-dioxygenase (α-DOX). The products of α-DOX decompose nonenzymatically into CO2 and the shortened aldehyde derivatives or, alternatively, are reduced either chemically by (i.e.) glutathione or enzymatically by the action of a peroxygenase (PXG). In contrast, the hydroperoxy products formed by LOXes have been shown to be relatively stable and are further metabolized by other enzymes such as special members of the group of P450 enzymes, the Cyp74-family: allene oxide synthase (AOS), hydroperoxide lyase (HPL) and divinylether synthase (DES), PXG or epoxyalcohol synthases (EAS) as illustrated in Figure 1 [1].
Lipases
In Arabidopsis several lipases have been described that supply the oxylipin pathway with fatty acid substrate. DEFECTIVE IN ANTHER DEHISCENCE1 (DAD1) [3] and DONGEL (DGL) [4] have been shown to belong to the family of phospholipases, which primarily may hydrolyze glycerophospholipids at the sn-1 position, but both enzymes may use glycolipids as substrates as well. On the basis of bioinformatic studies, it has been shown that both enzymes contain a putative N-terminal peptide sequence, a characteristic G-X-S-X-G motif and a catalytic triad (S, D and H). Recently, another DAD1-like lipase was identified in Arabidopsis as PLA-Iγ1. In contrast to DAD1 and DGL, this enzyme contributes to the early wound response of the plant and therefore might be involved in the release of preformed jasmonates from arabidopsides [4]. In addition, pPLA-I, a so-called patatin-related lipase, was also shown to play a role in regulating basal (but not pathogen-induced) production of jasmonates [5].
α-Dioxygenase (α-DOX)
α-DOX catalyzes the oxygenation of fatty acids at the C-2 atom by abstraction of the proR-hydrogen and stereoselective insertion of molecular oxygen yielding the respective 2R-hydroperoxy derivative [6]. The resulting compound is very unstable (with a half-life time (T1/2) of approximately 3 min at 23°C) and decomposes nonenzymatically into CO2 and the corresponding chain-shortened fatty aldehyde. Alternatively the hydroperoxy fatty acid may be reduced to corresponding 2-hydroxy fatty acid. Interestingly, α-DOX shows high homology towards both mammalian prostaglandin-synthase isoforms (PGHS-1 and PGHS-2) and both enzymes share common catalytic features [7]. Both enzymes contain heme as a co-factor that is crucial for catalytic activity. In both cases it is bound via two distinct His-residues that are placed on the distal and proximal site of the heme plane. Furthermore, both enzymes use a highly conserved tyrosine residue for the catalytic activity [8,9]. During the initiation process of the reaction a radical may be formed at this particular tyrosine, which in turn stereoselectively abstracts hydrogen from the respective C-atom of the fatty acid substrate yielding a carbon centred radical (Fig. 2).
Figure 2. Enantioselective dioxygenation of a long-chain fatty acid by α-DOX.
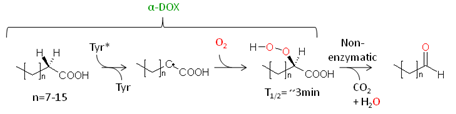
Molecular oxygen may then attack this radical, and a fatty acid hydroperoxyl radical is formed. It has been additionally shown that the Cα-H cleavage displays the first irreversible and turnover controlling step of the reaction. Due to a large weakly temperature-dependent kinetic isotope effect that was observed for this step, it is proposed that the homolysis of the C-H bond accomplished by quantum-mechanical nuclear tunnelling [10].
Lipoxygenase (LOX)
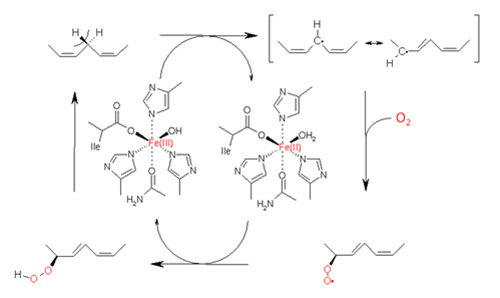
Plant LOX is a monomeric non-heme iron enzyme with a molecular weight of 90-110 kDa consisting of two different protein domains. The N-terminal region of approximately 25-30 kDa consists of a β-barrel domain with a yet unknown function. The C-terminal part of the enzyme harbours the catalytic domain, which is primarily formed by α-helices and has a molecular weight of approximately 55-65 kDa. The catalytic iron in the active site of a LOX is coordinated via 5 amino acid residues and a water molecule as the 6th ligand (Fig. 3). In a plant LOX, the iron is complexed via the N-atoms of three conserved histidines, the O-atom of one asparagine and the carboxy terminus of the C-terminal isoleucine [11].
Figure 3. Reaction cycle of LOX catalyzing the dioxygenation of a polyunsaturated fatty acid.
While most LOXes have been reported to catalyze dioxygenase reactions (i.e. bis-oxygenation of fatty acids) there are also a few LOXes known catalysing secondary conversions of hydroperoxy fatty acids and yielding epoxy-(alcohol)-derivatives [12] or lyase products [13].
The dioxygenase reaction of the LOX activity is characterized by five different key events mainly that are discussed in Figure 3:
- Enzyme activation
- Hydrogen abstraction
- Radical rearrangement
- Oxygen insertion
- Peroxyl radical reduction
The catalytic mononuclear iron centre of LOX enzyme exists in two different oxidation states – the ferrous (Fe(II)) and the ferric (Fe(III)). The resting enzyme contains ferrous iron and is inactive with any fatty acid substrate. However, upon reaction with hydroperoxides the iron becomes oxidized yielding ferric iron. This process known as the LOX activation is a single turnover process in which the once oxidized enzyme does not react with any hydroperoxide. The iron redox state cycles during catalysis between the inactive Fe(II)-H2O and the active Fe(III)-OH [14].
Fe(III)-OH functions as a base that stereospecifically abstracts a bisallylic pro-S hydrogen of a pentadiene system. It is now established that this reaction occurs via a proton-coupled electron transfer (PECT) mechanism in which the electron and the proton are transferred simultaneously from the substrate to the product. This reaction step is rate limiting and is also accompanied by a very large and weak temperature-dependent deuterium kinetic isotope effect. Thus, hydrogen abstraction is thought to follow a quantum-mechanical tunnelling mechanism in which the electron is not located on the proton but tunnels from the fatty acid to the catalytic Fe(III)-centre directly [15].
The abstraction of a bisallylic hydrogen leads to the formation of a carbon-centred radical that is delocalized via π-type electron system. Thus oxygen can be introduced at two different positions on the fatty acid backbone, either position [+2] or [-2]. In considering linoleic acid as substrate, where hydrogen abstraction takes place at C-11, the two positions for oxygen incorporation would be at C-9 or C-13 leading to the formation of 9-hydroperoxy- or 13-hydroperoxy derivatives, respectively (Fig. 4).
Figure 4. The regio- and stereospecific formation of linoleic acid hydroperoxides during LOX reaction.
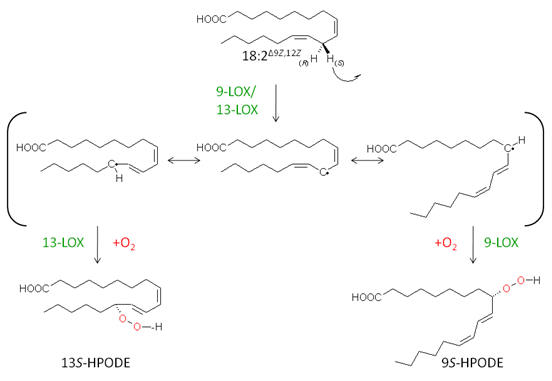
Although much effort has been made to explain the structural basis for the regiospecificity of LOX enzymes, until now there is no general unifying concept for this purpose. However, over the last two decades different critical amino acid residues have been identified, which determine the regiospecificity of LOX enzymes probably by shielding a particular position at the fatty acid backbone from molecular oxygen attack (the so-called space-related model). Another model that can explain the regiospecificity of LOX enzymes is the so-called substrate orientation model. According to this hypothesis the site of specific oxygen insertion is dependent of the substrate orientation within the active site of the LOX. In the case of, for example, linoleate 13-LOXes the fatty acid penetrates the active site with the methyl-end ahead, while for linoleate 9-LOXes an inverse substrate orientation modus is proposed where the fatty acid enters the enzyme with the carboxy-terminus first (“head-to-tail orientation”) [16].
On the other hand, oxygen insertion occurs always stereospecifically either leading to an R- or S-isomer. In all LOX enzymes analyzed so far, this specificity is mainly due to one particular amino acid residue in the active site of the enzyme – the so-called “Coffa-site”. While in S-specific LOX enzymes an alanine residue is located at this position, R-specific LOX enzymes contain a glycine residue.
Upon reaction with dioxygen a fatty acid peroxyl radical is formed that is highly reactive and can undergo secondary transformations like β-fragmentation, cis to trans double bond isomerisation or 5-exo-cyclisation. In order to circumvent these unspecific site reactions, the fatty acid peroxyl radical is efficiently trapped by reduction, yielding the corresponding hydroperoxy fatty acid derivative [17]. In the case of LOX enzymes, it is proposed that the peroxyl radical is reduced by accepting an electron from the catalytic iron centre via a PECT-mechanism and thus reactivating the enzyme.
Cyp74 Enzymes
The conversion of LOX-derived hydroperoxides displays a branching point in the metabolic pathway that is mainly catalyzed by enzymes belonging to the Cyp74 family, namely AOS, HPL, DES and EAS [18]. The Cyp74 family itself belongs to the superfamily of cytochrome P450 (P450s) enzymes that contain a cysteine-thiolate coordinated heme as cofactor and show a characteristic absorption maximum of 450 nm upon reduction and upon binding of CO. Common P450s catalyze monoxygenation reactions via a mechanism that involves binding of oxygen to the heme iron, sequential reduction, protonation of intermediary complexes and dissociation of water. Hence external electron donors are essential for the activity of classical P450-dependent monoxygenases [19]. In contrast to these classical P450 enzymes, the enzymes of the Cyp74 family do not require molecular oxygen and an external electron donor (e.g. NAD(P)H-dependent cytochrome-P450 reductase) for their catalytic activity as they use acyl hydroperoxides as substrate as well as electron donor. Furthermore, these enzymes catalyze isomerisation rather than monoxygenation reactions. As a consequence it is proposed that these types of enzymes use a short-circuited catalytic reaction cycle, which resembles that of classical P450s [20].
These functional differences of Cyp74 enzymes and classical P450 enzymes are also reflected by structural discrepancies in the active site architecture that preclude monoxygenase reactions in Cyp74s [21]:
- The signature heme binding loop of Cyp74 enzymes, that harbours the 5th heme-iron ligand, the conserved cysteine, has an additional nine amino acid insertion. This unique structural modification alters the active site topology and also the enzyme surface area that is known to be responsible for redox partner interactions.
- An important threonine residue that is in classical P450 enzymes crucial for generation of the highly activated iron-oxo species (compound I) is replaced by isoleucine or other hydrophobic alkane residues.
- The amide group of an essential asparagine residue is located directly above the heme group in a way that it can form hydrogen bonds with the hydroperoxy group of the substrate.
As shown in the Figure 5, the reaction mechanism of Cyp74 enzymes starts with the homolytic cleavage of the O-O-bond by oxidizing the heme centre to an iron-oxo species known as compound II from classical P450 chemistry. The alkoxyl radical produced undergoes cyclisation yielding an epoxy allylic radical derivative that displays the branching point in Cyp74-reaction. In the case of the AOS reaction, this intermediate is oxidized by compound II yielding an epoxy allylic carbocation derivative from which, upon elimination of the β-proton, the allene oxide is formed. In an alternative reaction, the epoxy C-C bond is cleaved resulting in the formation of a vinyl ether cation. This intermediate is either deprotonated, yielding the divinyl ether product (DES-reaction), or it is attacked by OH- at the cationic C-atom, yielding a hemiacetal derivative. The latter is unstable and decomposes into a short-chain aldehyde and an ω-oxo fatty acid (HPL-reaction). On the other hand, the above-mentioned allylic radical derivative can also rearrange by cleavage of the epoxy C-C bond forming a vinyl ether radical derivative. Upon reaction with compound II the hemiacetal derivative is formed via a classical oxygen rebound mechanism. Notably, this hemiacetal derivative is identical to the one formed via the “cationic pathway” and decomposes yielding the same products [20].
Figure 5. Reaction pathways of Cyp74 enzymes involving cationic or radical intermediates.
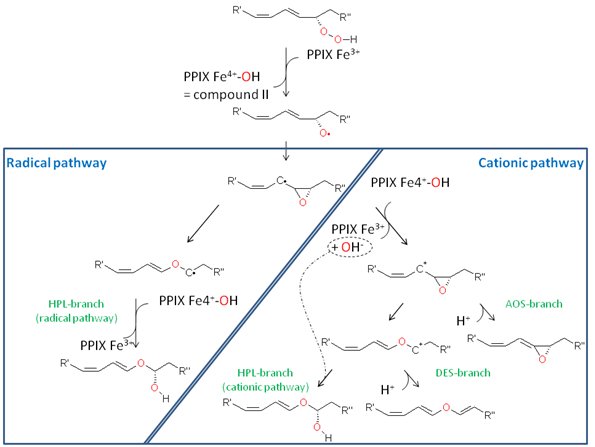
The close interconnection of the catalytic pathways of AOS and HPL is also mirrored by the fact that a single amino acid residue in the active site of the Cyp74-enzymes determines the respective catalytic activity, i.e. AOS activity or HPL activity. This amino acid residue is positioned in the active site of the enzyme facing the substrate carbon atom on which the centred radical or the positive charge is formed. In the case of AOS, the particular amino acid residue is a conserved phenylalanine that is thought to stabilize the carbon-centred radical via its aromatic site chain (cation-π interactions) and thus promoting oxidation of the radical by compound II resulting in allene oxide formation. On the other hand, in HPL the phenylalanine is replaced by a leucine. This residue cannot stabilize the carbon-centred substrate radical and thus no electron transfer to compound II can occur. Consequently, this leads to the formation of a vinyl-ether radical that is further oxidized by compound II to the hemiacetal derivative and thus this leads to the formation of HPL products [21].
Allene Oxide Cyclase
The allylic allene oxide formed by the AOS and 13-HPOTE (vide supra) has a very short half-time in water and is, in the absence of AOC, nonenzymatically converted to α- and γ-ketols or cyclized to racemic mixture of both cis(+)- and cis(-)-12-oxo phytodienoic acid (OPDA)-enantiomers. In the presence of AOC, however, the allene oxide is transferred towards the enantiomeric cis(+)-OPDA in a coupled reaction with the AOS [22,23].
The AOC is a homotrimeric enzyme with a molecular weight of approximately 20-25 kDa per subunit and belongs to the lipocalin protein family. Each subunit consists mainly of an eight-stranded anti-parallel β-barrel domain that forms a central hydrophobic cavity or barrel in which the reaction takes place. Notably, this region of the protein is very rigid and upon substrate binding no induced fit mechanism takes place [24]. Once the substrate is bound in the active site, one particular glutamate residue induces a partial charge separation leading to a delocalization of the Δ15-double bond. Thus, a positive charge is formed that is delocalized between C-13 and C-16 and opens the epoxy-ring. The oxyanion formed is stabilized via hydrogen bonding by one particular water molecule, which itself is tightly bound in a polar cavity formed by asparagines and a serine residue. In order to facilitate the ring closure that will form the cyclopentanone moiety, a conformational reorganisation from the cis to trans geometry around the C-10 – C-11 bond is necessary as shown in Figure 6. Notably, due to the steric limitations within the active site, the cis-trans isomerisation around the C-10 – C-11 bond leads to a further conformational change of the C-8 – C-9 bond which is also transferred from the cis- to the trans-geometry. The subsequent cyclisation is performed according to the rules of Woodward and Hoffmann [25,26].
Figure 6. Schematic overview of the reaction of AOC with 12,13(S)-epoxy-9(Z),11,15(Z)-octadecatrienoic acid
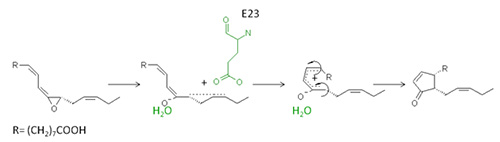
The tendency of the allene oxide to cyclise spontaneously in water yielding an enantiomeric mixture of OPDA points to the low energy barrier of this reaction. Thus, in terms of lowering this energy, AOC does not need much of an enzyme. It rather seems that the spontaneous cyclisation reaction proceeds in a stereoselective way that is controlled by steric active site determinants [26].
12-Oxo Phytodienoate Reductases (OPR)
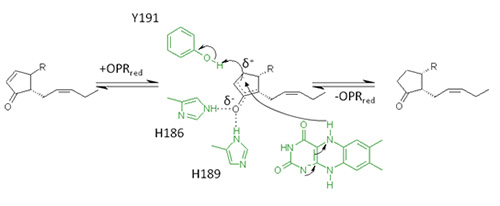
In plants 12S,13S- or cis(+)-OPDA is reduced to the corresponding cyclopentanone derivative by an enzyme known as OPDA reductase isoenzyme 3 (OPR3). This enzyme is a FMN-dependent oxidoreductase and belongs to the old yellow enzyme family – enzymes that are generally known to reduce double bonds of α,β-unsaturated carbonyls. OPR enzymes consist of an (α/β)8 barrel fold, in which eight α-helices surround eight parallel β-sheets forming a cylindrical architecture. The FMN cofactor is located in the centre of the enzyme, where it is noncovalently attached to the end of one particular β-barrel. Above the cofactor substrate specific amino acids are forming the active site cavity of the enzyme, in which the carbonyl-group of the substrate is attached and oriented via two strong hydrogen bonds to a His/His pair. In addition to substrate binding,this His/His pair is responsible for activating the α,β-double bond for hydride transfer to the β-C-atom of the substrate by polarizing it. The reduction of OPDA takes place via a so-called ping-pong bi-bi mechanism: In a first reaction step the FMN-cofactor is reduced by NAD(P)H followed by the release of NAD(P)+ (Fig. 7). In a second step OPDA binds to the enzyme and is reduced via hydride transfer to the β-C-atom [27-29].
Figure 7. Reaction mechanism for the reduction of 9S,13S-OPDA by OPR3.
Peroxygenase (PXG)
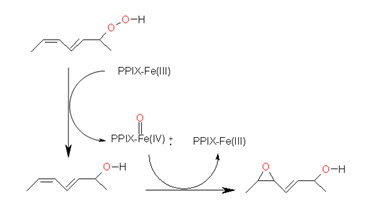
A further enzyme that uses LOX-derived fatty acid hydroperoxides as substrate is PXG. This enzyme catalyzes the reduction of a hydroperoxide derivative to the respective alcohol by transferring an oxygen atom to a substrate which in turn will be oxidized [30]. Like the members of the Cyp74 family this enzyme is also membrane bound in planta and contains heme as cofactor leading first to the misassumption that PXG also belongs to this enzyme family. However, in contrast to the Cyp74 enzymes that contain a thiolate-coordinated heme, in PXG, heme is bound via a conserved histidine residue that serves as the axial ligand. Its reaction is summarized in Figure 8.
Figure 8. Reaction pathway for the sequential reduction and oxidation of a fatty acid hydroperoxide by PXG yielding the corresponding fatty acid hydroxide as intermediate and an epoxy alcohol as end product.
Interestingly, PXG was shown to belong to the caloleosin family [31]. These enzymes are associated with oil bodies or lipid droplets via a hydrophobic central domain. This domain forms a hairpin-like structure that is deeply embedded into the lipophilic core of the droplet. The C- and N-terminal protein regions are located on the surface of the droplet and contain a putative protein kinase phosphorylation site at the C-terminus and a calcium binding EF-hand motif at the N-terminus.
Concluding Remarks
During last decades, several enzymes involved in plant oxylipin metabolism have been identified and characterized. Elucidation of some their 3D-structures as well as biochemical and biophysical studies on the respective reaction mechanisms not only contributed to the current status of a general understanding of enzymatic catalysis but also shed light on the evolutionary history of some of these enzymes. In addition to the enzymes showing “classical” catalytic activity (as they are mainly described in this review), further enzymes have been identified in recent years with an unusual or even an additional catalytic activity. In order to fully understand the mechanisms of oxylipin formation, it will be an important task to analyze these enzymes further.
References
- Andreou, A., Brodhun, F. and Feussner, I. Biosynthesis of oxylipins in non-mammals. Prog. Lipid Res., 48, 148-170 (2009) (DOI: 10.1016/j.plipres.2009.02.002).
- Howe, G.A. and Jander, G. Plant immunity to insect herbivores. Annu. Rev. Plant Biol., 59, 41-66 (2008) (DOI: 10.1146/annurev.arplant.59.032607.092825).
- Ishiguro, S., Kawai-Oda, A., Ueda, J., Nishida, I. and Okada, K. The DEFECTIVE IN ANTHER DEHISCENCE gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell, 13, 2191-2209 (2001).
- Ellinger, D., Stingl, N., Kubigsteltig, I.I., Bals, T., Juenger, M., Pollmann, S., Berger, S., Schuenemann, D. and Mueller, M.J. DONGLE and DEFECTIVE IN ANTHER DEHISCENCE1 lipases are not essential for wound- and pathogen-induced jasmonate biosynthesis: Redundant lipases contribute to jasmonate formation. Plant Physiol., 153, 114-127 (2010) (DOI: 10.1104/pp.110.155093).
- Scherer, G.F., Ryu, S.B., Wang, X., Matos, A.R. and Heitz, T. Patatin-related phospholipase A: nomenclature, subfamilies and functions in plants. Trends Plant Sci., 15, 693-700 (2010) (DOI: 10.1016/j.tplants.2010.09.005).
- Hamberg, M., Sanz, A. and Castresana, C. Fatty acid α-dioxygenases. Prostaglandins Other Lipid Mediat., 68-69, 363-374 (2002) (DOI: 10.1016/S0090-6980(02)00040-0).
- Koszelak-Rosenblum, M., Krol, A.C., Simmons, D.M., Goulah, C.C., Wroblewski, L. and Malkowski, M.G. His-311 and Arg-559 are key residues involved in fatty acid oxygenation in pathogen-inducible oxygenase. J. Biol. Chem., 283, 24962-24971 (2008) (DOI: 10.1074/jbc.M804358200).
- Smith, W.L., DeWitt, D.L. and Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem., 69, 145-182 (2000).
- Koeduka, T., Matsui, K., Akakabe, Y. and Kajiwara, T. Catalytic properties of rice α-oxygenase. A comparison with mammalian prostaglandin H synthases. J. Biol. Chem., 277, 22648-22655 (2002) (DOI: 10.1074/jbc.M110420200).
- Gupta, A., Mukherjee, A., Matsui, K. and Roth, J.P. Evidence for protein radical-mediated nuclear tunneling in fatty acid α-oxygenase. J. Am. Chem. Soc., 130, 11274-11275 (2008) (DOI: 10.1021/ja8042273).
- Liavonchanka, A. and Feussner, I. Lipoxygenases: Occurrence, functions and catalysis. J. Plant Physiol., 163, 348-357 (2006) (DOI: 10.1016/j.jplph.2005.11.006).
- Yu, Z., Schneider, C., Boeglin, W.E., Marnett, L.J. and Brash, A.R. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc. Natl. Acad Sci. USA, 100, 9162–9167 (2003) (DOI: 10.1073/pnas.1633612100).
- Senger, T., Wichard, T., Kunze, S., Göbel, C., Lerchl, J., Pohnert, G. and Feussner, I. A multifunctional lipoxygenase with fatty acid hydroperoxide cleaving activity from the moss Physcomitrella patens. J. Biol. Chem., 280, 7588-7596 (2005) (DOI: 10.1074/jbc.M411738200).
- Ivanov, I., Heydeck, D., Hofheinz, K., Roffeis, J., O'Donnell, V.B., Kühn, H. and Walther, M. Molecular enzymology of lipoxygenases. Arch. Biochem. Biophys., 503, 161-174 (2010) (DOI: 10.1016/j.abb.2010.08.016).
- Klinman, J.P. The role of tunneling in enzyme catalysis of C-H activation. Biochim. Biophys. Acta, 1757, 981-987 (2006) (DOI: 10.1016/j.bbabio.2005.12.004).
- Andreou, A. and Feussner, I. Lipoxygenases - Structure and reaction mechanism. Phytochemistry, 70, 1504-1510 (2009) (DOI: 10.1016/j.phytochem.2009.05.008).
- Schneider, C., Pratt, D.A., Porter, N.A. and Brash, A.R. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem. Biol., 14, 473-488 (2007) (DOI: 10.1016/j.chembiol.2007.04.007).
- Stumpe, M. and Feussner, I. Formation of oxylipins by CYP74 enzymes. Phytochem. Rev., 5, 347-357 (2006) (DOI: 10.1007/s11101-006-9038-9).
- Denisov, I.G., Makris, T.M., Sligar, S.G. and Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev., 105, 2253-2278 (2005) (DOI: 10.1021/cr0307143).
- Brash, A.R. Mechanistic aspects of CYP74 allene oxide synthases and related cytochrome P450 enzymes. Phytochemistry, 70, 1522-1531 (2009) (DOI: 10.1016/j.phytochem.2009.08.005).
- Lee, D.-S., Nioche, P., Hamberg, M. and Raman, C.S. Structural insights into the evolutionary paths of oxylipin biosynthetic enzymes. Nature, 455, 363-368 (2008) (DOI: 10.1038/nature07307).
- Hamberg, M. Biosynthesis of 12-oxo-10,15(Z)-phytodienoic acid: identification of an allene oxide cyclase. Biochem. Biophys. Res. Comm., 156, 543-550 (1988) (DOI: 10.1016/S0006-291X(88)80876-3).
- Brash, A.R., Baertschi, S.W., Ingram, C.D. and Harris, T.M. Isolation and characterization of natural allene oxides: unstable intermediates in the metabolism of lipid hydroperoxides. Proc. Natl. Acad. Sci. USA, 85, 3382-3386 (1988).
- Hofmann, E., Zerbe, P. and Schaller, F. The crystal structure of Arabidopsis thaliana allene oxide cyclase: Insights into the oxylipin cyclization reaction. Plant Cell, 18, 3201-3217 (2006) (DOI: 10.1105/tpc.106.043984 ).
- Schaller, F., Zerbe, P., Reinbothe, S., Reinbothe, C., Hofmann, E. and Pollmann, S. The allene oxide cyclase family of Arabidopsis thaliana - localization and cyclization. FEBS J., 275, 2428-2441 (2008) (DOI: 10.1111/j.1742-4658.2008.06388.x).
- Schaller, A. and Stintzi, A. Enzymes in jasmonate biosynthesis - Structure, function, regulation. Phytochemistry, 70, 1532-1538 (2009) (DOI: 10.1016/j.phytochem.2009.07.032).
- Breithaupt, C., Kurzbauer, R., Lilie, H., Schaller, A., Strassner, J., Huber, R., Macheroux, P. and Clausen, T. Crystal structure of 12-oxophytodienoate reductase 3 from tomato: Self-inhibition by dimerization. Proc. Natl. Acad. Sci. USA, 103, 14337-14342 (2006) (DOI: 10.1073/pnas.0606603103).
- Breithaupt, C., Kurzbauer, R., Schaller, F., Stintzi, A., Schaller, A., Huber, R., Macheroux, P. and Clausen, T. Structural basis of substrate specificity of plant 12-oxophytodienoate reductases. J. Mol. Biol., 392, 1266-1277 (2009) (DOI: 10.1016/j.jmb.2009.07.087).
- Breithaupt, C., Strassner, J., Breitinger, U., Huber, R., Macheroux, P., Schaller, A. and Clausen, T. X-ray structure of 12-oxophytodienoate reductase 1 provides structural insight into substrate binding and specificity within the family of OYE. Structure, 9, 419-429 (2001) (DOI: 10.1016/S0969-2126(01)00602-5).
- Blée, E. Biosynthesis of phytooxylipins: the peroxygenase pathway. Fett/Lipid, 100, 121-127 (1998).
- Hanano, A., Burcklen, M., Flenet, M., Ivancich, A., Louwagie, M., Garin, J. and Blée, E. Plant seed peroxygenase is an original heme-oxygenase with an EF-hand calcium binding motif. J. Biol. Chem., 281, 33140-33151 (2006) (DOI: 10.1074/jbc.M605395200).
In This Section
- Plant Fatty Acid Synthesis
- Production of Unusual Fatty Acids in Plants
- Arabidopsis Acyl-Coenzyme A-Binding Proteins
- Long Chain acyl-coA Synthetases and Other Acyl Activating Enzymes
- Plant Triacylglycerol Synthesis
- Triacylglycerol Biosynthesis in Eukaryotic Microalgae
- Subcellular Oil Droplets and Oleosins in Plants
- Triacylglycerol Mobilisation in Plants
- Role of Transcription Factors in Storage Lipid Accumulation in Plants
- Biosynthesis of Plant Lipid Polyesters
- Rubber Biosynthesis in Plants
- Carotenoid Biosynthesis and Regulation in Plants
- The Oxylipin Biosynthetic Pathways in Plants
- N-Acylphosphatidylethanolamines (NAPEs), N-acylethanolamines (NAEs) and Other Acylamides: Metabolism, Occurrence and Functions in Plants
- Phosphoinositide Signaling in Plants
- Plant Lipidomics
- 50 years of Galactolipid Research: The Beginnings
- Transport and function of lipids in the plant phloem