Fatty Acid beta-Oxidation
The Authors: Natasha Fillmore, Osama Abo Alrob and Gary D. Lopaschuk. The authors are from Cardiovascular Research Centre, Mazankowski Alberta Heart Institute University of Alberta, Edmonton, Canada. DOI: 10.21748/lipidlibrary.39187
Overview
Fatty acid β-oxidation is a multistep process by which fatty acids are broken down by various tissues to produce energy. Fatty acids primarily enter a cell via fatty acid protein transporters on the cell surface [1]. Fatty acid transporters include fatty acid translocase (FAT/CD36), tissue specific fatty acid transport proteins (FATP), and plasma membrane bound fatty acid binding protein (FABPpm) [1]. Once inside the cell, a CoA group is added to the fatty acid by fatty acyl-CoA synthase (FACS), forming long-chain acyl-CoA. Carnitine palmitoyltransferase 1 (CPT1) conversion of the long-chain acyl-CoA to long-chain acylcarnitine allows the fatty acid moiety to be transported across the inner mitochondrial membrane via carnitine translocase (CAT), which exchanges long-chain acylcarnitines for carnitine. An inner mitochondrial membrane CPT2 then converts the long-chain acylcarnitine back to long-chain acyl-CoA. The long-chain acyl-CoA enters the fatty acid β-oxidation pathway, which results in the production of one acetyl-CoA from each cycle of fatty acid β-oxidation. This acetyl-CoA then enters the mitochondrial tricarboxylic acid (TCA) cycle. The NADH and FADH2 produced by both fatty acid β-oxidation and the TCA cycle are used by the electron transport chain to produce ATP. An overview of fatty acid oxidation is provided in Figure 1.
Figure 1. Fatty Acid Oxidation Overview
Fatty acid β-oxidation is the process by which fatty acids are broken down to produce energy. Fatty acids primarily enter a cell via fatty acid protein transporters on the cell surface. Once inside, FACS adds a CoA group to the fatty acid. CPT1 then converts the long-chain acyl-CoA to long-chain acylcarnitine. The fatty acid moiety is transported by CAT across the inner mitochondrial membrane. CPT2 then converts the long-chain acylcarnitine back to long-chain acyl-CoA. The long-chain acyl-CoA can then enter the fatty acid β-oxidation pathway, resulting in the production of one acetyl-CoA from each cycle of β-oxidation. This acetyl-CoA then enters the TCA cycle. The NADH and FADH2 produced by both β-oxidation and the TCA cycle are used by the electron transport chain to produce ATP.

Role of Fatty Acid Supply in Regulating Fatty Acid β-Oxidation
Cellular fatty acid transport:
There has been considerable effort in recent years to elucidate the mechanisms by which the fatty acids are taken up by cells, particularly determining whether fatty acids are transported across the cellular membrane by simple diffusion or whether this transport is facilitated by membrane-associated proteins. While various results support both methods of transport, transport by membrane-associated proteins is believed to be the predominant means of fatty acid uptake into cells [2]. Various membrane proteins that facilitate cellular fatty acid uptake have been identified. The membrane-associated fatty acid transporters CD36/FAT, FABPpm, and FATPs differ in their molecular weight and degree of post-translational modification [3]. Stremmel et al. reported that antibodies directed against rat liver FABPpm inhibit fatty acid uptake by hepatocytes, adipocytes, and cardiomyocytes by 50-75%. This result suggested that a significant portion of fatty acid uptake is dependent on protein-mediated transport in different kinds of cells [3].
Schaffer and Lodish discovered FATP by use of an expression cloning strategy in adipocytes [4]. The expression of this 63 kDa integral membrane protein in a stable fibroblast cell line resulted in a 3-4 fold increase in long-chain fatty acid transport. Identification of this first FATP led to the discovery of several other isoforms of FATP (FATP1-6) [4]. FATP1 is predominantly expressed in heart and skeletal muscles [5]. FATP2 and FATP5 are expressed mainly in liver, where they are involved in hepatic lipid metabolism in association with acyl-CoA synthetase. FATP4 is essential for absorption of dietary lipids and has a critical role in normal skin structure and function. To date, FATP3 and FATP6 have been shown to have little or no fatty acid transport function [5].
The role of the membrane protein CD36/FAT in fatty acid uptake and β-oxidation in mammals has been studied extensively. CD36/FAT is an 88 kDa fatty acid translocase membrane protein expressed in proportion to the rate of fatty acid oxidation in muscle tissue (for example, it is expressed more in the heart than in skeletal muscle) [2]. CD36/FAT is involved in angiogenesis and inflammation, as well as lipid metabolism. Transfection of fibroblasts with CD36/FAT resulted in increased rates of fatty acid uptake [6]. Unlike FATP, CD36/FAT has the ability to translocate between intracellular endosomes and the plasma membrane of cells, which enables CD36/FAT to play a critical role in fatty acid uptake regulation. Insulin and muscular contraction can stimulate CD36/FAT translocation from intracellular stores to the plasma membrane, which leads to an enhanced uptake and β-oxidation of fatty acids. FoxO1 activation can lead to CD36/FAT translocation which results in increased fatty acid β-oxidation along with triacylglycerol accumulation [2]. This suggests a significant role of intracellular signaling in CD36/FAT function.
The exact mechanism inducing CD36/FAT translocation is still unknown. However, it is assumed that AMP-activated protein kinase (AMPK) activation and the energy status of muscle cells may participate in the CD36/FAT translocation response [7]. Furthermore, stimulation of protein kinase C (PKC) in cardiomyocytes induces CD36/FAT translocation, indicating a possible role for calcium in the translocation process [7]. On the other hand, inhibition of extracellular signaling receptor kinase (ERK) can block muscle contraction induced CD36/FAT translocation [7]. Post-translational modification of CD36/FAT through ubiquitination can also regulate the intracellular protein levels of CD36/FAT by targeting the protein for degradation. Therefore, insulin increases the availability of CD36/FAT for translocation by inhibition of ubiquitination. However, fatty acids promote ubiquitination, which leads to CD36/FAT degradation [1].
Fatty acid esterification to acyl-CoA:
A fatty acid must be converted to fatty acyl-CoA in order for it to enter the mitochondria and be oxidized [1]. The enzyme responsible for esterification of fatty acids to long-chain fatty acyl-CoA is FACS. For this reaction, FACS consumes the equivalent of two ATP. Another enzyme, cytosolic thioesterase (CTE), can remove the CoA converting the fatty acyl-CoA back to a fatty acid. Fatty acyl-CoA can either be converted to acyl carnitine, allowing it to be transported into the mitochondria and enter fatty acid β-oxidation or be converted to lipid metabolites (triacylglycerol, diacylglycerol, ceramide, etc.).
The acetyl-CoA carboxylase, malonyl-CoA decarboxylase, malonyl-CoA axis:
Acetyl-CoA carboxylase (ACC) is a central enzyme involved in fatty acid β-oxidation and fatty acid biosynthesis. ACC catalyzes the carboxylation of acetyl-CoA producing malonyl-CoA, which can be used by fatty acid synthase for fatty acid biosynthesis [1]. While malonyl-CoA is used as a substrate for fatty acid biosynthesis, malonyl-CoA is also a potent inhibitor of mitochondrial fatty acid uptake secondary to inhibition of CPT1 (Figure 2) [1]. There are two forms of ACC, a 265 kDa ACC1 isoform, which is highly expressed in the liver and adipose tissue, and a 280 kDa ACC2 isoform which is more specific to highly metabolic organs such as skeletal muscle and the heart [1]. AMPK plays a major role in ACC1 and ACC2 regulation by phosphorylating and inhibiting ACC activity. In situations of increased energy demand, AMPK is activated, where it then phosphorylates and inactivates both isoforms of ACC (Figure 3). ACC2 inhibition can lead to an increase in fatty acid β-oxidation, while fatty acid biosynthesis decreases when ACC1 is inhibited [1].
Figure 2. The fatty acid β-oxidation pathway
The four main enzymes involved in β-oxidation are: acyl-CoA dehydrogenase, enoyl-CoA hydratase, hydroxy acyl-CoA dehydrogenase, and ketoacyl-CoA thiolase. Acyl-CoA dehydrogenase creates a double bond between the second and third carbons down from the CoA group on acyl-CoA and in the process produces a FADH2. Next, enoyl-CoA hydratase removes the double bond just formed, in the process of adding a hydroxyl group to the third carbon down from the CoA group and a hydrogen on the second carbon down from the CoA group. Hydroxyacyl-CoA dehydrogenase removes the hydrogen in the hydroxyl group just attached and in the process produces a NADH. In the final step, ketoacyl-CoA thiolase attaches a CoA group on to the third carbon down from the CoA group resulting in the formation of two molecules, an acetyl-CoA and an acyl-CoA that is two carbons shorter.

Long-term regulation of ACC depends on regulation of its gene expression. Several transcriptional factors can regulate ACC gene expression, including sterol regulatory element binding protein (SREBP1a and SREBP1c) and carbohydrate response element binding protein (ChREBP) [8]. SREBP is regulated by insulin, which promotes the endoplasmic reticulum SREBP1c to be cleaved and translocated to the nucleus, leading to stimulation of ACC expression. Moreover, the transcription factors peroxisome proliferator activated receptor γ coactivator 1 (PGC-1) α and β can stimulate the expression of SREBP1a and SREBP1c, both of which have a vital role in lipogenesis. ChREBP expression can be induced by high glucose concentrations, resulting in the activated ChREBP promoting the expression of ACC1 and fatty acid synthase [8,9]. Nuclear respiratory factor-1 (NRF-1) is a principal modulator of mitochondrial protein expression and mitochondrial biogenesis, both of which are important for higher mitochondrial fatty acid β-oxidation capacity [10]. For example, Adam et al. showed that NRF-1 overexpression results in inhibition of ACC2 gene promoter activity in the mammalian heart, which enhances mitochondrial fatty acid β-oxidation rates, thereby promoting overall intracellular energetic capacity [10].
Malonyl-CoA decarboxylase (MCD) is the enzyme responsible for decarboxylation of malonyl-CoA to acetyl-CoA [1]. Generally, the level of malonyl-CoA is decreased when MCD activity is increased, resulting in an elevated rate of fatty acid oxidation. It has been reported that protein kinases that phosphorylate and inhibit ACC might activate MCD [1]. However, MCD appears to be primarily regulated by transcriptional means (discussed later). Therefore, MCD and ACC appear to work in harmony to regulate the pool of malonyl-CoA that can inhibit CPT1 [1].
Mitochondrial carnitine palmitoyl transferase (CPT):
The CPT isoform, CPT1, resides on the inner surface of the outer mitochondrial membrane and is a major site of regulation of mitochondrial fatty acid uptake [1]. As mentioned, CPT1 is potently inhibited by malonyl-CoA, the product of ACC that binds to the cytosolic side of CPT1. Mammals express three isoforms of CPT1, which are encoded by different genes. The liver isoform (CPT1α), the muscle isoform (CPT1β), and a third isoform of CPT1 (CPT1c), which is primarily expressed in the brain and testis [9]. More specifically, the heart expresses two isoforms of CPT1, an 82 KDa (CPT1α) isoform and the predominant 88 KDa (CPT1β) isoform (that has the highest sensitivity to malonyl-CoA inhibition). Insulin and thyroid hormone can regulate the sensitivity of CPT1α in the liver; however, the CPT1β isoform is not affected [9]. Previous studies have reported that the levels of malonyl-CoA are inversely correlated with fatty acid β-oxidation rates [1]. Furthermore, studies on ACC2 knockout mice suggest two separate cellular malonyl-CoA pools, malonyl-CoA produced by ACC1 (used mainly for lipogenesis), and a cytosolic pool of malonyl-CoA produced by ACC2 involved in the regulation of CPT1 and fatty acid β-oxidation [9].
Mitochondrial Fatty Acid β-Oxidation
The fatty acid β-oxidation pathway:
Fatty acid β-oxidation is the process of breaking down a long-chain acyl-CoA molecule to acetyl-CoA molecules. The number of acetyl-CoA produced depends upon the carbon length of the fatty acid being oxidized. This process involves a variety of enzymes, with the four main enzymes involved in fatty acid β-oxidation being, in order, acyl-CoA dehydrogenase, enoyl-CoA hydratase, hydroxyacyl-CoA dehydrogenase, and ketoacyl-CoA thiolase (Figure 3) [11]. At the end of each β-oxidation cycle, two new molecules are formed, an acetyl-CoA and an acyl-CoA that is two carbons shorter. Additionally, during β-oxidation NADH and FADH2 are formed. One FADH2 is produced during the reaction catalyzed by acyl-CoA dehydrogenase. An NADH is produced during the reaction catalyzed by hydroxyacyl-CoA dehydrogenase. The FADH2 and NADH produced during the process of fatty acid β-oxidation are used by the electron transport chain to produce ATP. There are different isoforms of these enzymes of β-oxidation, which have different affinities for different fatty acid chain lengths. For example, there is a very-long-chain acyl-CoA dehydrogenase, a long-chain acyl-CoA dehydrogenase, a medium-chain acyl-CoA dehydrogenase, and a short-chain acyl-CoA dehydrogenase. Interestingly, the enoyl-CoA hydratase, hydroxyacyl-CoA dehydrogenase, and ketoacyl-CoA isoforms specific for long-chain fatty acids form an enzyme complex on the inner mitochondrial membrane.
Figure 3. Key regulation sites of fatty acid β-oxidation
Fatty acid β-oxidation is regulated at multiple levels. This figure shows some of the ways fatty acid β-oxidation is regulated. 1. Regulation can occur at the level of fatty acid entry into the cell. AMPK, PKC, and PPARγ positively regulate the activity of CD36/FATP. 2. Regulation also occurs via the regulation of the levels of acetyl-CoA and malonyl-CoA. AMPK inhibits ACC, resulting in increased acetyl-CoA levels/decreased malonyl-CoA levels and increased fatty acid oxidation. Malonyl-CoA inhibits fatty acid oxidation by inhibiting CPT1. 3. Transcriptional regulation is also involved in regulating fatty acid β-oxidation. PGC-1α, a transcription factor coregulator, and the transcription factor PPARα act in the nucleus to increase transcription of mitochondrial genes, fatty acid utilization genes, and other transcription factors.
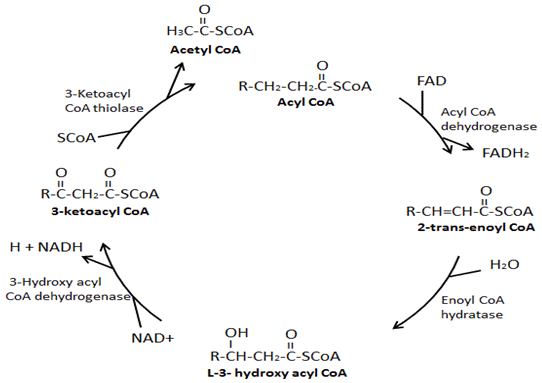
Auxiliary enzymes are required for the β-oxidation of unsaturated fatty acids and odd-chain fatty acids. Odd-numbered fatty acids are broken down by β-oxidation to acetyl-CoA molecules and propionyl-CoA. While propionyl-CoA could be metabolized through alternative pathways, it is primarily metabolized in the cell to succinyl-CoA by three enzymes (propionyl-CoA carboxylase, methylmalonyl-CoA epimerase, and methylmalonyl-CoA mutase) [11-14]. This succinyl-CoA can then enter the TCA cycle. Compared to even-numbered fatty acids, odd-numbered fatty acids occur infrequently in nature [15]. The two auxiliary enzymes, enoyl-CoA isomerase and 2,4-dienoyl-CoA reductase are necessary for the complete oxidation of unsaturated fatty acids [11]. During the β-oxidation cycle in which the cis-double bond begins on the third carbon of the acyl-CoA, the first step involves enoyl-CoA isomerase isomerizing it before enoyl-CoA hydratase, and the other two enzymes, can act on the acyl-CoA. A double bond on an even-numbered carbon requires both the auxiliary enzymes. Once the double bond is on the fourth carbon of the acyl-CoA at the beginning of a β-oxidation cycle it begins to be oxidized. Following action of acyl-CoA dehydrogenase, 2,4-dienoyl CoA reductase acts on the acyl-CoA followed by enoyl-CoA isomerase. Enoyl-CoA hydratase then acts on the acyl-CoA and the process resumes its normal order.
Allosteric control of fatty acid β-oxidation:
The activity of the enzymes of fatty acid β-oxidation is affected by the level of the products of their reactions [16]. Each of the β-oxidation enzymes is inhibited by the specific fatty acyl-CoA intermediate it produces [17]. Interestingly, 3-ketoacyl-CoA can also inhibit enoyl-CoA hydratase and acyl-CoA dehydrogenase [17]. β-Oxidation can also be allosterically regulated by the ratio of NADH/NAD+ and acetyl-CoA/CoA level. A rise in the NADH/NAD+ or acetyl-CoA/CoA ratios results in inhibition of fatty acid β-oxidation. Increases in the acetyl-CoA/CoA ratio have specifically been shown to lead to feedback inhibition of ketoacyl-CoA thiolase [16].
Fatty acid β-oxidation can also occur in peroxisomes. In animals, peroxisomes are believed to be important in the initial breakdown of very-long-chain fatty acids and methyl branched fatty acids [11]. The enzymes involved in fatty acid oxidation in peroxisomes are different from mitochondria. An important difference is acyl-CoA oxidase, the first enzyme in peroxisome β-oxidation, which transfers the hydrogen to oxygen producing H2O2 instead of producing FADH2. The H2O2 is broken down to water by catalase. Importantly, the fatty acyl-CoA intermediates formed during β-oxidation are the same in peroxisomes and mitochondria. Peroxisomes also contain the necessary enzymes for α-oxidation, which are necessary for oxidation of some fatty acids with methyl branches.
Transcriptional regulation of fatty acid β-oxidation:
The proteins involved in fatty acid β-oxidation are regulated by both transcriptional and post-transcriptional mechanisms. There are a number of transcription factors that regulate the expression of these proteins. The peroxisome proliferator-activated receptors (PPARs) and a transcription factor coactivator PGC-1α are the most well known transcriptional regulators of fatty acid β-oxidation [18]. PPARs and Retinoid X receptor heterodimerize and bind to gene promoters containing the PPAR response element [18]. Examples of proteins involved in fatty acid β-oxidation that are transcriptionally regulated by the PPARs include FATP, acyl-CoA synthetase (ACS), CD36/FAT, MCD, CPT1, long-chain acyl-CoA dehydrogenase (LCAD), and medium-chain acyl-CoA dehydrogenase (MCAD) [18]. Estrogen-related receptor α (ERRα) has also been implicated in the regulation of fatty acid β-oxidation, having been shown to also regulate transcription of the gene encoding MCAD [18]. Ligands that bind to and modulate the activity of PPARα, δ, and γ include fatty acids [18].
The genes regulated by each of the PPARs vary between tissue types. For example, skeletal muscle PPARδ, but not PPARα, upregulates expression of CPT1 [19]. PPAR isoforms are also differentially expressed between tissue types [18]. While PPARδ protein tends to be ubiquitously expressed, PPARα is predominantly expressed in highly metabolic tissues (i.e. heart, skeletal muscle, and liver) and PPARγ is predominantly expressed in tissues such as adipose tissue [18]. Until recently, PPARγ was not believed to play a significant role in regulating fatty acid β-oxidation. However, recent knockout and overexpression studies have suggested that PPARγ may have a role in regulating fatty acid β-oxidation. Over expressing PPARγ in cardiac muscle results in increased mRNA levels for fatty acid β-oxidation proteins [20].
The transcriptional co-activator PGC-1α binds to and increases the activity of PPARs and ERRα to regulate fatty acid β-oxidation [21]. PGC-1α modulates the activity of a number of transcription factors that can increase the expression of proteins involved in fatty acid β-oxidation, the TCA cycle, and the electron transport chain. For example, increasing PGC-1α protein expression induces massive mitochondrial biogenesis in skeletal muscle [21].
PGC-1α is regulated at both the gene and protein level. AMPK increases the activity of pre-existing PGC-1α protein through two proposed mechanisms. The first is by phosphorylating PGC-1α on threonine and serine residues results in an overall increase of the PGC-1α activity [22]. AMPK may also increase the activity of PGC-1α by activating sirtuin 1 (SIRT1). SIRT1 can then deacetylate PGC-1α, increasing its activity [22]. It is believed that AMPK increases PGC-1α mRNA levels by regulating the binding of transcription factors to specific sequences located in the PGC-1α gene promoter which include two MEF sites, one cAMP response element (CRE) site, and the GATA/Ebox region [22]. AMPK regulates the MEF sites by phosphorylating GEF, a protein which can mediate movement of MEF2 into the nucleus [22]. AMPK may increase binding to the CRE site by phosphorylation of cAMP-response element binding protein (CREB) 1 and other members of the CREB family that bind to CRE promoter regions [22]. As another example, free fatty acids can also regulate PGC-1α protein expression. For instance, a high-fat diet can elevate levels of PGC-1α in rat skeletal muscle [23].
Conclusions
Fatty acid β-oxidation is major metabolic pathway that is responsible for the mitochondrial breakdown of long-chain acyl-CoA to acetyl-CoA. This process involves many steps that are regulated at the transcriptional and post-transcriptional level. Transcriptional regulation involves PPARs, SREBP1, and PGC-1α, while the post-transcriptional level mainly involves allosteric control of fatty acid β–oxidation, as well as ACC, MCD, and CPT regulation. Both mechanisms work in harmony to ensure a continual supply of long-chain acyl-CoA for β-oxidation, and products of β-oxidation for mitochondrial energy production.
References
- Lopaschuk, G.D., Ussher, J.R., Folmes, C.D., Jaswal, J.S. and Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol Rev., 90, 207-258 (2010).
- Su, X. and Abumrad, N.A. Cellular fatty acid uptake: a pathway under construction.Trends Endocrinol. Metab., 20, 72-77 (2009).
- Glatz, J.F., Luiken, J.J. and Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev., 90, 367-417 (2010).
- Folmes, C.D. and Lopaschuk, G.D. Regulation of fatty acid oxidation of the heart. In: Mitochondria: The Dynamic Organelle (1st Edition), pp. 27-62 (S.W. Schaffer and M.S. Suleiman (eds.), Springer, New York) (2007).
- Nickerson, J.G., Momken, I., Benton, C.R., Lally, J., Holloway, G.P., Han, X.X., Glatz, J.F., Chabowski, A., Luiken, J.J. and Bonen, A. Protein-mediated fatty acid uptake: regulation by contraction, AMP-activated protein kinase, and endocrine signals. Appl. Physiol. Nutr. Metab., 32, 865-873 (2007).
- Bonen, A., Chabowski, A., Luiken, J.J. and Glatz, J.F. Is membrane transport of FFA mediated by lipid, protein, or both? Mechanisms and regulation of protein-mediated cellular fatty acid uptake: molecular, biochemical, and physiological evidence. Physiology (Bethesda), 22, 15-29 (2007).
- Holloway, G.P., Luiken, J.J., Glatz, J.F., Spriet, L.L. and Bonen, A. Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: an overview. Acta Physiol. (Oxford), 194, 293-309 (2008).
- Tong, L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci., 62, 1784-1803 (2005).
- Schreurs, M., Kuipers, F. and van der Leij, F.R. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev., 11, 380-388 (2010).
- Adam, T., Opie, L.H. and Essop, M.F. AMPK activation represses the human gene promoter of the cardiac isoform of acetyl-CoA carboxylase: Role of nuclear respiratory factor-1. Biochem. Biophys. Res. Commun., 398, 495-499 (2010).
- Schulz, H. Oxidation of fatty acids in eukaryotes. In: Biochemistry of Lipids, Lipoproteins and Membranes (5th Edition), pp. 131-154 (D.E. Vance and J. Vance (eds.), Elsevier, Amsterdam) (2008).
- Mazumder, R., Sasakawa, T. and Ochoa, S. Metabolism of propionic acid in animal tissues. X. Methylmalonyl co-enzyme A mutase holoenzyme. J. Biol. Chem., 238, 50-53 (1963).
- Kaziro, Y., Leone, E. and Ochoa, S. Biotin and propionyl carboxylase. Proc. Natl. Acad. Sci. USA, 46, 1319-1327 (1960).
- Beck, W.S., Flavin, M. and Ochoa, S. Metabolism of propionic acid in animal tissues. III. Formation of succinate. J. Biol. Chem., 229, 997-1010 (1957).
- Diedrich, M. and Henschel, K.P. The natural occurrence of unusual fatty-acids. 1. Odd numbered fatty-acids. Nahrung, 34, 935-943 (1990).
- Schulz, H. Regulation of fatty acid oxidation in heart. J. Nutr., 124, 165-171 (1994).
- Eaton, S. Control of mitochondrial β-oxidation flux. Prog. Lipid Res., 41, 197-239 (2002).
- Huss, J.M. and Kelly, D.P. Nuclear receptor signaling and cardiac energetics. Circ. Res., 95, 568-578 (2004).
- Dressel, U., Allen, T.L., Pippal, J.B., Rohde, P.R., Lau, P. and Muscat, G.E. The peroxisome proliferator-activated receptor β/δ agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol., 17, 2477-2493 (2003).
- Son, N.H., Park, T.S., Yamashita, H., Yokoyama, M., Huggins, L.A., Okajima, K., Homma, S., Szabolcs, M.J., Huang, L.S. and Goldberg, I.J. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J. Clin. Invest., 117, 2791-2801 (2007).
- Lin, J., Handschin, C. and Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell. Metab., 1, 361-370 (2005).
- Jensen, T.E., Wojtaszewski, J.F. and Richter, E.A. AMP-activated protein kinase in contraction regulation of skeletal muscle metabolism: necessary and/or sufficient?Acta Physiol. (Oxford), 196, 155-174 (2009).
- Hancock, C.R., Han, D.H., Chen, M., Terada, S., Yasuda, T., Wright, D.C. and Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA, 105, 7815-7820 (2008).
Acknowledgements: GDL is a Scientist of the Alberta Heritage Foundation for Medical Research
In This Section
- Glycerophosphate and Acylglycerophosphate Acyltransferases
- Mammalian Diacylglycerol Acyltransferases (DGAT)
- Fatty Acid beta-Oxidation
- Regulation of Lipins and Their Role in Lipid Metabolism
- Phospholipid Biosynthesis
- Phospholipases
- Acylglycerol Lipases (Neutral Lipid Hydrolysis)
- Metabolism and Function of Very-Long-Chain Polyunsaturated Fatty Acids (>C24) in Mammals