The Chromatographic Resolution of Chiral Lipids
The Author: William W. Christie, James Hutton Institute (and Mylnefield Lipid Analysis), Invergowrie, Dundee (DD2 5DA), Scotland.
The following was first published by W.W. Christie, in Advances in Lipid Methodology - One, pp. 121-148 (1992) (Ed. W.W. Christie, Oily Press, Ayr), and it is now reproduced here by kind permission of P.J. Barnes & Associates (The Oily Press), who retain the copyright. This review is now substantially out of date in terms of applications and chromatographic media, but no comparable review has been published recently and it remains a useful guide to the early literature. A comprehensive list of recent references to the topic can be accessed here.
- Introduction
- Resolution of Chiral Glycerol Compounds
1. Separation of diastereomeric derivatives by HPLC on silica gel
2. Resolution by HPLC on chiral stationary phases
3. Resolution by gas chromatography - Resolution of Chiral Oxygenated Fatty Acids including Eicosanoids
1. Resolution by gas chromatography
2. Separation of diastereomeric derivatives by HPLC on silica gel
3. Resolution by HPLC on chiral stationary phases
4. Resolution by thin-layer and other chromatographic procedures - Resolution of Chiral Branched-Chain Fatty Acids
- Some Miscellaneous Separations
- References
Introduction
Any molecule has the property of chirality or asymmetry when its mirror image cannot be superimposed on itself. The two non-superimposable forms are termed enantiomers or optical isomers, and have the same chemical bonds but differ in the arrangement of these bonds in space and thus in their three-dimensional structure; they cannot be interconverted without breaking and reforming one of the bonds. This is illustrated with reference to glyceraldehyde, first chosen arbitrarily by Fischer as the reference standard, in Figure 1.
Figure 1. The absolute configuration of glyceraldehyde enantiomers and their designation.

Enantiomers of a specific compound will rotate the plane of polarization of polarized light by equal amounts but in different directions. When they interact with reagents that are non-chiral, they do so in exactly the same way since they have identical chemical properties, although any products formed will have different molecular configurations. Their physical properties, such as melting points, solubilities and partition coefficients, are also identical. On the other hand, each enantiomer may react at a different rate with a given enantiomeric form of a chiral reagent. In order to effect the resolution of enantiomers by chromatographic means, therefore, it is necessary that they be made to interact with chiral molecules in either the mobile or the stationary phase of whatever system is adopted.
Molecules with more than one centre of asymmetry can exist in 2n forms, where n is the number of asymmetric centres. Stereoisomers of such molecules in which all the corresponding asymmetric centres are inverted are enantiomers; those in which the configuration of one or more (but not all) of the asymmetric centres differs are termed diastereomers. With a given compound, diastereomers may have slightly different conformations and for example small changes in intra-molecular hydrogen bonding and thus different internal energies. They have distinct chemical and physical properties and thus have the potential to be separated in non-chiral chromatographic environments.
Lipids like all natural compounds are synthesised by enzyme systems which are themselves chiral and which invariably produce chiral products. The asymmetry may be inserted directly during the biosynthesis of the aliphatic chains of lipids, for example in isoprenoid and other branched-chain lipids where the precursors are themselves chiral. Alternatively, the chirality may be introduced to the aliphatic chain later as in the synthesis of oxygenated or cyclopropane fatty acids. The nature and the absolute configurations of chiral fatty acids, wax components and sphingoid bases have been reviewed by Smith [125]. As with other natural compounds, the Cahn-Ingold-Prelog system for designating absolute configurations is followed and the chiral centres are designated R (rectus) or S (sinister) according to the approved rules of priority of each substituent. This sometimes has the unfortunate consequence that changes in the R or S designation may be required with eicosanoids on the addition or removal of a double bond during its metabolism, for example. The older D/L nomenclature is still encountered in the literature and may be used when the absolute configuration is uncertain.
The glycerol moiety of glycerolipids also has a centre of asymmetry at the second carbon atom (Fig. 2). For convenience, a "stereospecific numbering system", based on the R/S nomenclature, is favoured since it reflects the true stereochemistry of such molecules and simplifies their naming [69,70]. In a Fischer projection of a natural (S)-glycerol derivative, the secondary hydroxyl group is shown to the left of C-2; the carbon atom above this is C-1 and that below is C-3. The prefix "sn" is placed before the stem name of the compound. Smith [126] reviewed the topic of glyceride chirality.
Figure 2. A Fischer projection of a triacyl-sn-glycerol to illustrate the stereospecific numbering system [69,70].

The primary method for the determination of the stereochemical configuration of a chiral molecule involves chemical synthesis from a precursor of defined stereochemistry by procedures that do not cause inversion of the asymmetric centres. Comparisons of the sign of the Cotton effect in the optical rotatory dispersion (ORD) curves, i.e. the plot of degree of rotation against wavelength of plane-polarized light, of unknowns with similar compounds of defined configuration can also lead to conclusive results. Increasingly nowadays, nuclear magnetic resonance (NMR) spectroscopy in the presence of chiral shift reagents is being used for the same purpose. However, the magnitude of the observed effects both with optical and NMR measurements for most natural lipids is generally too small to be of practical value.
Once the stereochemistry of the reaction of an enzyme is known from studies of the structures of a precursor and its product, the enzyme may be applied to other substrates to define their stereochemistry or to prepare a particular enantiomer. In recent years, the chromatographic behaviour of chiral compounds or their derivatives in suitable systems has begun to contribute to the solution of stereochemical problems. The development of methods for the chromatographic resolution of chiral compounds in general has also made it much easier to obtain the pure enantiomers that may be required for studies of biological activity. The pharmaceutical industry has been a major beneficiary and has stimulated much of the published work, but lipid analysts have been quick to take advantage of new opportunities.
Many factors have contributed to the possibilities for improved resolution of enantiomeric lipids by chromatographic means. These include technical developments such as the construction of the many components of high-performance liquid chromatography (HPLC) systems, including pumps, injection systems and detectors, which have lead to such enormous improvements in the capabilities and efficiency of this technique [34]. It is even possible to purchase an HPLC detector that senses optical activity in the eluent, though applications to lipids have yet to be described. In gas chromatography (GC), the advent of capillary columns of fused silica has been a major advance [35]. However, it may be argued that most important of all has been the knowledge gained of the effects of chirality on chromatographic selection mechanisms, especially in the laboratory of W.H. Pirkle. In particular, this has lead to the discovery of suitable derivatives for the resolution of diastereomers and to the development of novel stationary phases for HPLC with chiral moieties bonded chemically to inert supports. HPLC offers great opportunities in that there is flexibility in the choice of both the mobile and stationary phases to enhance resolution.
In 1979, Pirkle and House [119] first put chiral chromatography on a systematic basis with a theoretical explanation of the "three-point rule", i.e. there must be at least three simultaneous interactions between a chiral stationary phase and a solute enantiomer and one of these must be stereochemically dependent if chiral resolution is to be effected. Amongst the intermolecular forces that influence chiral resolution are hydrogen bonds, pi-pi, dipole-dipole, steric and hydrophobic interactions. The rule appears to hold for each instance in which the mechanism of chiral selectivity is known in detail and with many different types of chromatographic system. In effect, the solute and the stationary phase form a transient diastereomeric complex, and the configurations of the substituents in each enantiomer will determine the strengths of the interactions and the order of elution.
One advantageous strategy for effecting a separation, for example, can be to include a naphthyl or anthryl group (electron-rich) in the stationary phase to cause it to interact via the pi bonds with an electron-deficient aromatic moiety (e.g. dinitrophenyl) in the solute, perhaps after suitable derivatization of the latter. Similarly, the hydrogen atom of a secondary amide can interact strongly with the oxygen of a carboxyl group via hydrogen bonding. For optimum resolution, the bonds which interact should be as close as possible to the asymmetric carbon atoms.
The stereochemistry of organic molecules is a highly complex topic and the reader is referred to comprehensive texts for detailed information [71,121]. Similarly, the chromatographic resolution of chiral compounds in general has been the subject for substantial books [86,160]. The latter topic was reviewed in relation to fatty acids briefly in 1976 by Smith [127], to eicosanoids (by means of HPLC) by Brash and Hawkins [27] and to lipids in general by Takagi in 1990 [129]. The reader is referred to these and earlier reviews [125,126] for information on historical aspects of the chirality of lipids and for more detailed discussion of lipid stereochemistry per se. This review is concerned primarily with the newer chromatographic methods for the resolution of chiral lipids or for the determination of their stereochemical configurations.
Resolution of Chiral Glycerol Componds
1. Separation of diastereomeric derivatives by HPLC on silica gel
One of the more important stereochemical problems with glycerolipids is the resolution of diacyl-sn-glycerols, because of their importance as biosynthetic precursors of triacyl-sn-glycerols and glycerophospholipids. One approach to the resolution of diacylglycerols has been to react them with isocyanates prepared from enantiomerically pure amines to form diastereomeric urethane (or carbamate) derivatives of which the most useful have proved to be (S)-(+)- or (R)-(-)-1-(1-naphthyl)ethyl urethanes. Such diastereomers can then be resolved by adsorption chromatography. The required reagents (the corresponding isocyanates) are available commercially and reaction with alcohols occurs rapidly in the presence of a basic catalyst in toluene solution with no apparent racemization [93]. They are readily detected as they emerge from HPLC columns by their pronounced UV absorbance at 280 nm. Equally important, the parent alcohol is readily regenerated following resolution by reaction of the urethane derivative with trichlorosilane [38,115,117].
Diastereomeric naphthylethyl and other urethanes especially have been utilized for the resolution of alcohols and amines of many kinds by column chromatography on non-chiral adsorbents such as silica gel or alumina [7]. This is possible because diastereomers have different physical and chemical properties as discussed briefly above. Thus a chiral derivatizing agent (R)-X will react with an enantiomeric substance (R,S)-Y as -

In a given chromatographic system, the degree of separation obtained of the two diastereomers will depend on the chiral structures of X and Y and the manner of their interactions with the mobile and stationary phases. A further useful feature is that the order of elution of the diastereomeric derivatives is reversed in the same chromatographic system if the other enantiomer of the reagent, i.e. (S)-X, can be employed. It is of course essential that the derivatizing agent be of a high degree of optical purity, otherwise a further pair of diastereomers would be formed to contaminate the required compounds, and that derivatization should not be accompanied by significant racemization.
With diastereomeric naphthylethyl urethanes and related compounds, it appears that the presence of the hydrogen atom on the nitrogen atom between the chiral centres is crucial to the separation process and is presumed to be the primary binding site via hydrogen bonding to silanols on the adsorbent surface [116]. The amide bond is planar in nature and facilitates binding to the polar surface of the adsorbent. In determining the order of elution of the diastereomers, the magnitudes of the steric effects of the substituents on the chiral carbon atom of the urethane group and the first carbon of the alcohol moiety are then the important factors. The degree of resolvability or chromatographic diastereoselectivity is given by the separation factor (α value). As with resolution on chiral stationary phases, this is greatest when the chiral centres in the diastereomers are close together. Bulky or planar substituents such as a naphthyl moiety aid the separation by their contribution to the overall conformational rigidity of the molecule. In contrast to urethanes, diastereomeric ester derivatives tend to be poorly resolved by adsorption chromatography because there is virtually no hydrogen bonding to the surface.
Diastereomeric urethane derivatives were first used for the HPLC resolution of chiral glycerol compounds by Michelsen et al. [93]. They were able to obtain base-line resolution of (S)-1-(1-naphthyl)ethyl urethane derivatives of dialkyl-sn-glycerols, such as 2,3- and 1,2-O-dihexadecyl-sn-glycerols; the compounds eluted in the order stated from columns of silica gel with hexane-ethyl acetate (85:15, v/v) as the mobile phase. Reducing the content of ethyl acetate improved the resolution (increased the RS value), while lengthening the time of elution, but addition of a modifier such as isopropanol gave poorer results. bis-(S)-1-(1-Naphthyl)ethyl urethane derivatives of 3-O- and 1-O-hexadecyl-sn-glycerol were also well resolved. However, the practical value of the separations was limited, since it was only possible to effect them with compounds containing one type of alkyl group; partial separation of homologues caused groups of components of different stereochemistry to overlap when the method was applied to mixed substrates. In addition, partial resolution only of diastereomeric derivatives prepared from diacyl-sn-glycerols could be achieved under the conditions essayed. It was, however, determined that the derivatives gave distinctive mass spectral fragmentation patterns of value for characterization purposes.
In contrast, Laakso and Christie [84] were able to effect excellent resolution of diastereomeric naphthylethyl urethane derivatives of diacyl-sn-glyerols on silica gel as illustrated in Figure 3. Diacyl-sn-glycerols derivatized with (S)-(+)-1-(1-naphthyl)ethyl isocyanate eluted in the order 1,3-, followed by 1,2- and 2,3-isomers; Michelsen et al. [93] reported that the 1,2- and 2,3-dialkyl analogues eluted in the reverse order. In addition, molecular species of those single-acid diacyl-sn-glycerols derivatives were in general well resolved in the order 18:1 < 18:0 < 18:2 < 16:0, i.e. neither that expected for normal- nor reversed-phase partition chromatography. Presumably it is a reflection of the conformations of the fatty acids and their influence on one of the "three-point" interactions between the substrate and the adsorbent. When the (R)- form of the derivatizing agent was utilized, the elution order changed so that 2,3- emerged before 1,2-isomers as expected. It is noteworthy that with the common range of fatty acyl groups studied here, the 1,2-dipalmitoyl- and 2,3-dioleoyl-sn-glycerol urethanes were just separable when the (S)-form of the derivatives was employed, but these were furthest apart with the (R)-form. If the two forms of the derivatives were to be used in conjunction with HPLC-mass spectrometry, it should in theory be possible to analyse complex mixtures in some detail. The optimum resolution was achieved with a mobile phase consisting of hexane with a small amount of isopropanol as modifier. Little resolution was achieved with modifiers other than alcohols, in direct contradiction of the findings of Michelsen et al. [93]. Subsequently, it was shown that slightly better resolution and more reproducible retention times were obtainable with n-propanol (containing 2% water) in hexane or isooctane as the mobile phase [36].
Figure 3. Separation of a standard mixture of single acid 1,2- and 2,3-diacyl-sn-glycerols in the form of the chiral 1-(1-naphthyl)ethyl urethane derivatives by HPLC. S: (S)-(+)-1-(1-naphthyl)ethyl urethanes. R: (R)-(-)-1-(1-naphthyl)ethyl urethanes. Two columns of silica gel (Hypersil™ 3µm, 250 x 4.6 mm i.d.) in series were used with hexane-isopropanol (99.5:0.5, v/v) at a flow-rate of 0.8 mL/min as the mobile phase and UV detection at 280 nm. (Reproduced by kind permission of the authors and of Lipids [84]).

This type of resolution served as the basis for a new procedure for the stereospecific analysis of triacyl-sn-glycerols from natural fats and oils, i.e. for the determination of the compositions of the fatty acids esterified to each of positions sn-1, sn-2 and sn-3 [36,84]. The triacyl-sn-glycerols were first subjected to partial hydrolysis with ethyl magnesium bromide and the products were converted to the (S)-(+)-1-(1-naphthyl)ethyl urethane derivatives. The derivatized diacylglycerols were isolated by solid-phase extraction chromatography on octadecylsilyl columns and the diastereomeric forms were resolved by HPLC on silica gel. By determining the fatty acid compositions of the intact triacylglycerols and of the 1,2- and 2,3-diacyl-sn-glycerol derivatives, the compositions of positions sn-3 and sn-1, respectively, were calculated; that of position sn-2 was obtained by difference. Previously, this type of analysis could only be accomplished by time-consuming procedures involving further synthetic steps, stereospecific lipase hydrolyses and more complex separation procedures (reviewed elsewhere [30,33]). Some comparable approaches to the problem are described in the next section.
R-(+)-1-Phenylethyl urethane derivatives of diacylglycerols have also been resolved by HPLC with columns of silica gel in studies designed to determine the positional specificity of lipases [122]. Others reported that these derivatives were not as effective for the purpose as the analogous naphthylethyl urethanes [36].
2. Resolution by HPLC on chiral stationary phases
Great strides have been made in the resolution of chiral glycerolipids by HPLC since 1985, especially in the laboratory of Takagi and Itabashi in Japan. Their general approach has been to convert mono- and diacyl-sn-glycerols and related compounds to the 3,5-dinitrophenyl urethane (DNPU) derivatives for resolution by HPLC on columns containing a stationary phase with chiral moieties bonded chemically to a base of silica gel. In this instance also, the presence of the hydrogen atom on nitrogen in the urethane group and available for hydrogen bonding with the stationary phase (presumably to the carboxyl moieties) appears to be essential if resolution is to be achieved. The 3,5-dinitrophenyl moieties of the urethanes contribute to charge-transfer interactions with functional groups having pi electrons on the stationary phase. A further advantage of these urethane derivatives is that they exhibit appreciable absorbance in the UV spectrum over a wide range of wavelengths, although the optimum is at 226 nm [66].
Enantiomeric monoacylglycerols were first separated after conversion to the 3,5-dinitrophenyl urethanes on a column (250 × 4 mm i.d.) packed with a stationary phase consisting of (S)-2-(4-chlorophenyl)isovaleroyl-D-phenylglycine linked to aminopropyl-silanized silica (5 µm particles, Sumipax™ OA-2100) [66,135]. Base-line resolution of the derivative of 1-stearoyl-sn-glycerol from the sn-3 analogue (in this order) was obtained with a mobile phase of hexane-1,2-dichloroethane-ethanol (40:12:3 by volume) [135]. Subsequently with minor modifications to the mobile phase, improved resolutions were obtained and some separation of homologous compounds was achieved such that parallel linear relationships were observed between the logarithms of the retention volumes and the carbon numbers of the compounds for the sn-1 and sn-3 series; retention times decreased with increasing chain-length [66]. Overlap between the two series was seen when carbon numbers differed by six units. Comparable resolution of enantiomers of the analogous monoalkyl-sn-glycerols has been reported [136]. In addition, such compounds (and some dialkylglycerols) were resolved in the form of the 3,5-dinitrobenzoyl derivatives by HPLC on a stationary phase containing bonded D-naphthylalanine [32]. It appeared that the presence of the 3,5-dinitrobenzoyl moiety was mandatory for resolution to be achieved. However, benzyoylisopropylidene derivatives of glycerol were successfully resolved by HPLC on a column of Chiralcel OB™ [75].
Greatly improved separations of the urethane derivatives of monoacylglycerol enantiomers were reported when the stationary phase for HPLC contained N-(R)-1-(1-naphthyl)ethylaminocarbonyl-(S)-valine as the functional moiety bonded to silica gel (Sumipax™ OA-4100) [130]. By using a long column (500 × 4 mm i.d.), slow flow-rate (0.5 mL/min), mobile phase of low polarity (hexane-1,2-dichloroethane-ethanol, 40:10:1 by volume) and consequently very long retention times (six hours or more), base-line separations of homologues differing in total chain length by as many as ten carbons were achieved within each group of enantiomers. Similarly, C18 enantiomers with zero to three double bonds were each cleanly separated, though isomers such as the 16:0 and 18:1 compounds were critical pairs and tended to elute together. A decreasing linear relationship between the logarithms of the retention times (volumes) and carbon numbers was again observed, and with increasing numbers of double bonds in the molecules the logarithms of the retention times increased in a linear fashion. Recently this methodology was adapted to serve as the basis of an alternative procedure for stereospecific analysis of triacyl-sn-glycerols (see also Section B.1 above) [131]. 1 and 3-Monoacyl-sn-glycerols were prepared from triacylglycerols by partial hydrolysis with ethyl magnesium bromide; they were isolated by TLC on layers of silica gel impregnated with boric acid, separated into saturated and unsaturated fractions by silver ion chromatography, converted to the dinitrophenylurethane derivatives and then resolved on a Sumipax™ OA-2100 column. The positional distributions in each of positions sn-1, -2 and -3 could be calculated from the data. Later, an improved procedure was described in which a Sumichiral™ OA 4100 column was employed [8,132-134]. In this instance, there was no need for the step involving silver ion chromatography, since adequate resolution of complicated mixtures of 1- and 3-sn-glycerol derivatives was achievable. By lowering the column temperature and slowing down the flow-rate, the method could even be applied to such complex triacyl-sn-glycerols as fish oils. This procedure will certainly be widely used in the future.
Parallel separations of 3,5-dinitrophenyl urethane derivatives of chiral diacyl-sn-glycerols and structurally related compounds by HPLC on chiral phases have also been carried out. In the light of the resolutions achieved most recently, those of diacylglycerol derivatives obtained on the Sumipax™ 2100 column appear rather limited [67]. Nonetheless, clear resolution of single-acid 1,3-, 1,2- and 2,3-diacyl-sn-glycerols was achieved and straight-line relationships between the logarithms of the retention volumes and carbon numbers were observed for homologous series of each form. More valuable practical separations of these and analogous compounds were obtained on the Sumipax™ OA-4100 column, i.e. of mono-acid diacyl-, dialkyl- [137] and alkylacylglycerol [139] and di-acid diacylglycerol derivatives [41]. For optimum resolution as with the monacylglycerol derivatives described above, a long column (500 × 4 mm i.d.), slow flow-rate (0.25 mL/min) and mobile phase of low polarity (hexane-1,2-dichloroethane-ethanol, 170:10:1 by volume) were utilized [140]. Molecular species differing by two carbon atoms were separated to the base-line and distinct peaks were obtained for saturated diacyl-sn-glycerol homologues differing in total chain length by up to six carbon atoms, before overlap of the different enantiomeric groups was seen. It was even possible to resolve 1-alkyl-3-acyl-rac-glycerols [139].
It has been established that diacylglycerol derivatives separated by chiral-phase HPLC in this way give distinctive mass spectra which can be utilized for structure determination [83].
Appreciably better resolution was reported from the laboratories of Takagi and Kuksis with a column (250 × 4.6 mm i.d.) in which a polymer of (R)-(+)-1-(1-naphthyl)ethylamine moieties was bonded chemically to silica gel (YMC-Pack A-K03™) [64]. The elution order was the same as that with the previous columns, and the best enantiomer separations were achieved with mobile phases similar to those described above, e.g. hexane-1,2-dichloroethane-ethanol (40:10:1 by volume) at a flow-rate of 1 mL/min; the same types of molecular interaction are certainly involved. It is noteworthy that this column is capable of giving resolutions of great practical value with elution times and other chromatographic parameters in a more conventional range for HPLC. The method was applied to diacylglycerol derivatives generated from natural triacyl-sn-glycerols by partial hydrolysis with a Grignard reagent, and an application showing the resolution of molecular species within each of the 1,2- and 2,3-diacyl-sn-glycerol groups prepared from corn oil is illustrated in Figure 4. Species of 1,3-diacylglycerols were also well separated, though enantiomers were not resolved, but a preliminary separation by thin-layer chromatography (TLC) on boric acid-impregnated layers was desirable. The procedure clearly has considerable potential for the analysis of molecular species of diacylglycerols as part of structural analyses of triacyl-sn-glycerols and especially for stereospecific analysis of the latter (see previous section). Indeed, an application of this type has now appeared [159]; 1,2- and 2,3-diacyl-sn-glycerols, again generated by Grignard hydrolysis of triacyl-sn-glycerols, were resolved on a YMC-Pack A-KO3™ column as the dinitrophenyl urethane derivatives, and the compositions of positions sn-1, sn-2 and sn-3 were calculated by methods analogous to those described above. This is likely to be one of the major practical applications of the methodology. In combination with mass spectrometry especially, the elution system should be capable of application to the most complex natural samples, such as fish oils or butter fat.
Figure 4. Chiral phase HPLC resolution of the 1,2- and 2,3-diacyl-sn-glycerol moieties of corn oil triacylglycerols as 3,5-dinitrophenyl urethanes [64]. Peaks 1 to 3 represent 16:0-18:1 + 18:1-18:1, 16:0-18:2 + 18:1-18:2, and 18:2-18:2, respectively. Conditions are given in the text. (Reproduced by kind permission of the authors and of the Journal of Lipid Research, and redrawn from the original).

In addition, this chiral column was utilized to isolate 1,2- and 2,3-sn-diacylglycerols so that the molecular species of each could be determined by capillary GC on a polar stationary phase [65]. The approach was to resolve the diacylglycerols, generated by reaction of triacyl-sn-glycerols with ethyl magnesium bromide, in the form of the DNPU derivatives on the chiral column, then to regenerate the parent diacylglycerol molecules by reaction with trichlorosilane (with no acyl migration) and convert them to the trimethylsilyl ethers for the GC analysis.
Unfortunately, both the columns containing the required chiral stationary phases and the derivatizing reagent are costly and can only be purchased from sources in Japan (at the time this article was first written). When these are more readily available, this methodology will certainly be used more widely in structural studies of glycerolipids.
3. Resolution by gas chromatography
Near base-line resolution of diastereomeric alpha-methoxy-alpha-trifluoromethylphenylacetic acid derivatives of diethanoyl-sn-glycerols has been achieved by GC on a fused-silica capillary column coated with a methylsiloxane (SE-54™) stationary phase [94]. Unfortunately, it was not possible to resolve diacylglycerol derivatives of longer-chain fatty acids, and this together with the difficulty of preparing the derivatives is a limitation on the practical value of the procedure. Short-chain 1,2- and 1,3-dialkyl-sn-glycerols in the form of diastereomeric benzylethyl or naphthylethyl urethanes were resolved in a similar manner [128].
Resolution of Chiral Oxygenated Fatty Acids Including Eicosanoids
1. Resolution by gas chromatography
In 1966, the first published report of the resolution of diastereomeric fatty acid derivatives appeared by Wood et al. [157], who were able to separate trifluoroacetate derivatives of tri- and tetrahydroxystearates on columns packed with the non-polar silicone phase, SE-30™, or better the polar polyester, EGSS-X™. The absolute configuration of one of the hydroxyl groups was known, and the assignments of the others were made from their chromatographic behaviour on GC and TLC (see Section C.4 below) and their melting points.
Many different types of enantiomeric derivatives have been used to prepare diastereomers of hydroxy fatty acids for resolution by gas chromatography. The quality of the separation appears to depend both on the nature of the derivatizing reagent and on the position of the hydroxyl group in the aliphatic chain. In 1968, GC was first used to accomplish the resolution of methyl esters of 2-hydroxy acids (short-chain or aromatic) after they had been further converted to the diastereomeric L-(-)-menthyloxycarbonyl derivatives [152]. The same methodology was used subsequently by Hammarstrom [55,56] to confirm that the homologous 2-hydroxy fatty acids of cerebrosides have the D-configuration. Here also, the methyl esters of the fatty acids were converted to the (-)-menthylformates and separated on a packed column coated with the methyl silicone, SE-30™, as the stationary phase; the natural D-enantiomer eluted after the L-form. An improved procedure for the derivatization step was reported by others [10]. In order to improve the separation with 2- [22,49] and 3-, 4-, 3- and 2-hydroxy fatty acids [56,57], diastereomeric (R)-(+)-1-phenylethyl urethane and D-phenylpropionate derivatives, respectively, were introduced for GC analysis, with QF-1™ as the stationary phase. The latter derivative permitted much better resolution of 3- than of 2-isomers, i.e. the opposite of the result obtained with the menthyl formates, but no separation of diastereomeric 4-, 7- or 13-hydroxystearates was seen. However, near base-line resolution of D and L-17-hydroxystearates was attained following conversion to the urethane derivatives [22].
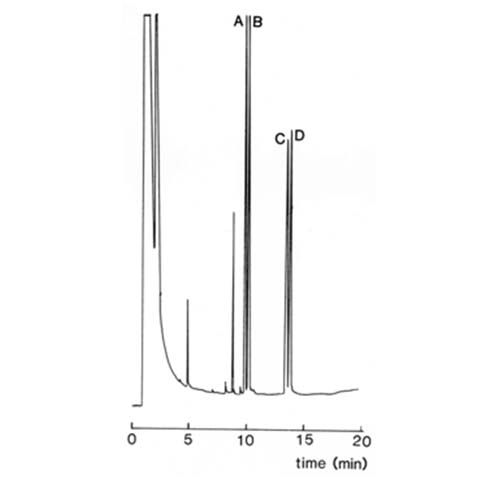
Greatly improved results are possible with modern fused-silica capillary columns, and base-line separations of the diastereomeric (-)-alpha-methoxy-alpha-trifluoromethylphenylacetate derivatives of methyl DL-2-hydroxy-palmitate and -stearate on a column of this type, with OV-1™ as the stationary phase, have been recorded as illustrated in Figure 5 [19].
Fig. 5. Separation of diastereomeric methoxytrifluoromethylphenyl acetate derivatives of methyl 2-hydroxypalmitate (A,B) and 2-hydroxystearate (C,D) by GC on fused silica columns (25 m × 0.32 mm]) coated with OV-1™ [19]. The column was temperature-programmed from 180 to 240°C at 2°C/min, and helium was the carrier gas. (Reproduced by kind permission of the authors and of the Journal of Chromatography, and redrawn from the original).
With a fatty acid containing a hydroxyl group located centrally, as in the hydroperoxides of linoleic acid, it was not possible to accomplish resolution of diastereomeric menthylformates by GC [50]. The proportions of the various enantiomeric products of soybean and corn lipoxygenase, i.e. 13-hydroperoxy-9,11-octadecadienoic and 9-hydroperoxy-10,12-octadecadienoic acids, were, however, determined by reducing them to the corresponding hydroxides and subjecting the menthylformate derivatives to oxidative ozonolysis; the resulting short-chain 2-menthyloxycarbonyl fatty acids were methylated and resolved by packed-column GC with QF-1™ as the stationary phase [50]. As the stereochemistry of the product was not changed by the sequence of reactions, that of the parent hydroperoxide was immediately obvious. The same procedure has been utilized to determine the stereochemistry of a number of hydroperoxides including eicosanoids [24,25,28,51, 54,59,88,90,92,153]. Following conversion to allylic alcohols, the absolute configurations of epoxides were obtained similarly [42], and this methodology in combination with other chromatographic techniques, especially complexation TLC with arsenite-containing layers, was used to determine the stereochemistry of trihydroxy acids derived from linoleic acid [52,53]. The problem of stereochemical assignments of hydroxyl groups was also tackled by a modified procedure in which acetylated hydroxy acids were cleaved by oxidative ozonolysis, converted to the (R)-(+)-2-butyl esters and then reacetylated for analysis by capillary GC [147-149].
12- and 13-Hydroxystearates (the latter derived from a linoleate hydroperoxide) in the form of the sesquiterpenoid drimanoyl derivatives were partially resolved by packed column GC with SE-30™ as the stationary phase [31]. Better resolution might be expected with modern capillary columns, but applications of such derivatives do not appear to have been pursued further.
As an alternative approach to the resolution of chiral hydroxy acids, a diastereomeric amide derivative of the carboxyl group with (+)-phenylethylamine was prepared for resolution by GC (and by TLC as discussed in Section C.4 below) [73]. A packed column with OV-1™ as the stationary phase was employed. Partial resolution was obtained with the diastereomeric hydroxy fatty amide, the derivative of the L-2-hydroxy acid eluting first, but near base-line separation was achieved when the hydroxyl group was methylated.
4- and 5-Hydroxy acids cyclize spontaneously to form gamma- and delta-lactones, respectively. These have been resolved by GC after conversion to diastereomeric dicarbamate [40] or carbamoyloxy carboxamide [41] derivatives on capillary columns coated with conventional stationary phases, and in underivatized form on a chiral (cyclodextrin-based) stationary phases [11,76].
2. Separation of diastereomeric derivatives by HPLC on silica gel
As with the glycerol derivatives discussed above (Section B.1), enantiomeric hydroxy acids and eicosanoids especially, following conversion to appropriate diastereomeric derivatives, have been resolved by HPLC on silica gel. Several types of derivative have been employed, but there appears to be no consensus as to which is best since commercial availability has imposed a restriction. For example, the dehydroabietyl urethane derivatives of an eicosanoid were first resolved by low-pressure column chromatography [38] (see Section E below), and have since been used in a number of HPLC applications. Thus Falck et al. [43] reduced a number of epoxyeicosatrienoic acids to hydroxy compounds, converted them to dehydroabietyl urethane derivatives and were able to effect base-line resolutions by HPLC on silica gel with mixtures of hexane and small amounts of isopropanol as the mobile phase; (R)-5-, 8-, 9-, 11-, 12- and 15-hydroxy-eicosatetraenoates eluted before the corresponding (S)-forms. Various hydroxy-eicosatetraenoic acids have been resolved in the same way [28,89,158].
Diastereomeric (R)-(-)-menthyloxycarbonyl derivatives of hydroxy-eicosatetraenoic acids have also been resolved by means of HPLC. In the first such report [37], a cation-exchange column (Nucleosil™ SA) in the silver ion form and methanol as the mobile phase were used to effect separation of 15-hydroxyeicosatetraenoate diastereomers, and a similar type of column was used by others for positional isomers of related compounds [59]. It is not clear how or indeed whether the silver ions participate in the separation in a significant manner, although one of the "three-point" interactions could be between silver ions and the double bond system adjacent to both chiral centres. Others have achieved similar resolutions on silica gel [17,29]. Such diastereomers were not resolved by reversed-phase HPLC, and this technique was used to isolate specific diastereomer mixtures prior to resolution.
(+)-alpha-Methoxy-alpha-trifluoromethylphenylacetate derivatives of hydroxy compounds derived from lipoxygenase-catalysed reaction with linoleic and arachidonic acids were resolved by HPLC on silica gel [9]. In contrast to most observations with such separations of diastereomers, ethyl acetate was found to be a more effective polar modifier of the mobile phase than was isopropanol. Derivatives from both 9- and 13-hydroxyoctadecadienoates were easily resolved, and adequate separation of diastereomers of 15-hydroxyeicosatetraenoate was achieved. The same method was subsequently employed to resolve the enantiomeric forms of 11-hydroxylauric acid [12].
Racemic hydroperoxides per se from polyunsaturated fatty acids have been resolved by preparing novel diastereomeric perketals, i.e. 2-propenyl ethers derived from trans-2-phenylcyclohexanol [114]. Many of the positional isomers examined were separable by HPLC in the adsorption mode on silica gel with hexane-ethyl acetate (99:1, v/v) as the mobile phase, generally with the hydroperoxide of the S-configuration preceding the R-form (Fig. 6). Only the 11- and 15-isomers derived from arachidonate could not be resolved by this means. More surprisingly, excellent resolution of diastereomers from all the positional isomers examined (5-, 6-, 8-, 9-, 11-, 15-) was obtained by reversed-phase HPLC, with an octadecylsilyl stationary phase and acetonitrile containing 0.1% triethylamine as the mobile phase. In this instance, hydroperoxides of the R-configuration always preceded the S-form. The ketal derivatives were readily hydrolysed under acidic conditions without racemization to the required hydroperoxides.
Figure 6. HPLC separation of perketal derivatives of hydroperoxides prepared from methyl arachidonate with a column of silica gel, hexane-ethyl acetate (99:1, v/v) as mobile phase and UV detection at 254 nm [114]. The positions and stereochemistry of the hydroperoxyl groups are indicated. (Reproduced by kind permission of the authors and of Chemical Research Toxicology, and redrawn from the original).

Diastereomeric (8S,15S)- and (8R,15S)-dihydroxyeicosatetranoates have been resolved by HPLC on columns of silica gel with hexane-2-propanol-acetic acid (100:3:0.1, by volume) or modified mixtures as the mobile phase [26,29,92]; the stereochemistry of the double bonds had some effect on retention times. These compounds have also been resolved by HPLC in the reversed-phase mode [48]. Similarly, diastereomeric 11,12- and 14,15-dihydroxyeicosatetraenoates have been resolved under comparable conditions [153,154]. With compounds such as this with two chiral carbons, no derivatization was necessary in theory or in practice to obtain the required resolution.
3. Resolution by HPLC on chiral stationary phases
Since 1987 when the first applications to lipids appeared, analysts have been turning increasingly to chiral stationary phases in order to resolve enantiomers of hydroxy fatty acids and hydroxy-eicosanoids especially by means of HPLC. Such columns are easy to use and can permit sensitive analysis. Also, better results are possible as the commercial stationary phases are made from materials of higher optical purity than are some derivatizing agents used in alternative procedures. Often there is no need to convert to derivatives that may have to be hydrolysed later to regenerate the compounds of interest, although resolution may be enhanced if this approach is taken. On the debit side, the columns are costly and may not be reproducible in their properties. Brash and Hawkins [27], for example, state in relation to columns of the type introduced by Pirkle that "there appears to be an element of luck involved in obtaining a "good" column". As the chiral moiety is linked to the silica by both ionic and covalent bonds, loss of resolution may become apparent after continued use through leaching of ionically bound species. Some manufacturers offer a regeneration service for columns that have deteriorated.
The most widely used column of this kind is one developed by W.H. Pirkle, i.e. (R)-(-)-N-3,5-dinitrobenzoylphenylglycine (DNBPG) bonded chemically to silica gel (Bakerbond™, Regis Hi-Chrom™). Such a column was used for example to completely resolve the enantiomeric forms of the hydroxy compounds derived from linoleic acid by the action of lipoxygenase (followed by reduction), i.e. 13- and 9-hydroxyoctadecadienoates (Fig. 7), and to partially resolve all the related compounds prepared from arachidonic acid in a single chromatographic run [82]. The carboxyl group was methylated but the hydroxyl group was not derivatized. With a mobile phase of hexane and 0.5% isopropanol, S- enantiomers eluted before the R-forms. Others have resolved enantiomeric hydroxyeicosanoids by the same procedure [13-16,23,44,46,59,77-81,113, 145,155,156], and as pentafluorobenzyl ester derivatives in order to assist with subsequent analyses by GC-mass spectrometry [144].
Figure 7. Chiral phase HPLC separation of reduced hydroperoxide derivatives from linoleic acid on a DNBPG column with hexane-isopropanol (99.5:0.5, v/v) as mobile phase and UV detection at 235 nm [82]. The positions and stereochemistry of the hydroxyl groups are indicated. (Reproduced by kind permission of the authors and of Analytical Biochemistry, and redrawn from the original).

The phenacyl ester derivative of the epoxy compound, 2-tetradecylglycidic acid, was resolved in this manner [150], as were related compounds with different substituents in position 2 or with the acid group derivatized in alternative ways [151]. When the temperature of the column and the mobile phase was reduced to 0 to 5°C, the resolution was improved appreciably.
Much better resolution of hydroxy-eicosanoids on columns of the DNBPG type was reported after conversion of the analytes to benzoyl or naphthoyl derivatives [60]. It is believed that increased interactions between the electron-deficient dinitrobenzoyl groups of the chiral phase and the electron-rich benzoyl/naphthoyl moieties of the solute facilitated the separation. (The separation principle is thus complementary to that of dinitrophenylurethane derivatives on a naphthoyl stationary phase as discussed in Section B.2 above). For a range of positional isomers, the order of elution was R before S, i.e. the opposite of that for the hydroxy esters. After isolation of such enantiomers by preparative HPLC, the original hydroxy acids could be recovered by mild alkaline hydrolysis without racemization. Again this methodology has been adopted by others for related compounds [36,63,112,146], including isomers which have the hydroxyl group in positions 18 and 19 and lack conjugated double bonds [112]. A systematic study of different derivatives and makes of HPLC column confirmed that naphthoyl derivatives gave the best results, but indicated that the method used to prepare the stationary phase also affected the resolution, ionically linked DNBPG groups giving the best results [80]. In addition, it was demonstrated that some diastereomeric dihydroxy-eicosanoids, not separable by HPLC with a column of silica gel, could be resolved on the chiral phase.
An impression is rapidly being obtained that hydroxy-eicosanoids with underivatized hydroxyl groups are most easily resolved on Chiralcel™ columns in which the stationary phase is cellulose containing covalently-bonded aromatic moieties. Kitamura et al. [74] used a Chiralcel™ OC (cellulose trisphenylcarbamate) column and hexane-isopropanol (99:1, v/v) to resolve racemic 12-hydroxyeicosatetraenoates, that of the R-configuration eluting before the S-form, and this was confirmed by others [87,141]. Excellent resolution of analogous 8- and 15-hydroxy compounds has also been achieved [27]. This type of column was used under similar conditions to resolve synthetic precursors of prostaglandins and a prostaglandin analogue [58,96], as was the naphthoyl derivative of 19-hydroxyeicosatetraenoate [112]. A Chiracel™ OB (cellulose trisbenzoate) column was utilized to resolve enantiomers of 8-hydroxyeicosatetraenoate, the S-form eluting before the R-form in this instance, as illustrated in Figure 8 [62].
Figure 8. HPLC resolution methyl 8S- and 8R-hydroxyeicosatetraenoate on a column (250 × 4.6 mm) of Chiralcel™ OB, with a mobile phase of hexane-isopropanol (100:2, v/v) at a flow rate of 0.5 mL/min and with UV detection at 235 nm [62]. (Reproduced by kind permission of the authors and of Biochimica Biophysica Acta, and redrawn from the original).

All the regioisomers of racemic epoxy-eicosatrienoates, as the pentafluorobenzyl esters, were completely resolved on columns of Chiralcel™ OB and OD (cellulose tris-3,5-dimethylphenyl-carbamate) [58,72]. With most of the isomers, the columns were used in the adsorption mode but separation of the enantiomeric 5,6-epoxy-eicosatrienoates was possible in the reversed-phase mode only, i.e. with a mobile phase of water-ethanol (3:7, v/v). When Chiralcel™ HPLC columns are used in the latter manner, great care is necessary to avoid high pressures as these can cause compaction of the stationary phase. Epoxy acids prepared from linoleic acid by reaction with soybean peroxygenase have also been resolved on a Chiralcel™ OB column [23].
A third type of chiral column, i.e. with N-(S)-2-(4-chlorophenyl)isovaleroyl-D-phenylglycine ionically bonded to silica and slurry-packed into a fused-silica capillary column, was utilized to effect resolution of dinitrophenyl urethane derivatives of 2-hydroxy acids [138]. Apart from the fact that particularly good resolution was achieved, this type of column made an economical use of the costly stationary phase.
A column containing a chiral glycoprotein phase (Chiral AGP™) has been used to resolve enantiomeric hydroxypentadienes related in structure to leukotrienes [120].
4. Resolution by thin-layer and other chromatographic procedures
Some remarkable separations of diastereomeric organic molecules have been achieved by adsorption chromatography on silica gel in thin layers or in low-pressure columns, rivaling those obtained by HPLC indeed in quality if not in convenience (c.f. references [115,116,118], for example). It may be that the high degree of hydration of the silica gel in these circumstances has a beneficial effect on the separation. Unfortunately, such methodology appears to have been tested to a limited extent only with lipids.
As with other chromatographic systems discussed above, resolution is effected most easily when the chiral centre is at position 2 of the fatty acid molecule. For example, the D and L forms of 2-hydroxypalmitic acid were each obtained from the racemic mixture by conversion to the methyl esters and then to the L-acetylmandelate derivatives by Hitchcock and Rose [61] (although they attribute the method to L.J. Morris and M.L. Crouchman who apparently did not publish it as intended). The diastereomers were clearly separated by TLC on layers of silica gel with benzene-diethyl ether (97:3, v/v) as the mobile phase; the lower band (Rf = 0.56) was D-(L-acetylmandelyloxy)palmitate and the upper (Rf = 0.72) was the corresponding L-form. The same method was used to determine the configuration of the 2-hydroxy acids from myelin [142] and from the wax esters of rat skin lipids [18]. Racemic 2-hydroxy-palmitic and -stearic acids as the (-)-alpha-methoxy-alpha-trifluoro-5-methylphenylacetates have been resolved by TLC on layers of silica gel modified with chemically bonded phenylmethylvinyl groups [19]. On the other hand, when there is a second chiral centre adjacent to the hydroxyl group, it has proved possible to achieve useful separations without derivatizing the latter [143]. Thus the racemic 2-hydroxy compound prepared from 3D,7D,11D-phytanic (3,7,11,15-tetramethylhexadecanoic) acid as the methyl ester was resolved into the diastereomeric forms by TLC on silica gel with hexane-ethyl acetate (9:1, v/v) as the mobile phase. Wood et al. [157] were able to resolve diastereomeric trihydroxy fatty acids, derived from ricinoleate, by TLC on silica gel impregnated with sodium arsenite and with chloroform-methanol (98:2, v/v) as the mobile phase. TLC procedures were also utilized by Morris and coworkers for the determination of the absolute configuration of a natural epoxy fatty acid, (+)-vernolic (cis-12,13-epoxy-cis-9-octadecenoic) acid and related compounds. The stereochemistry of the epoxyl group was correlated with that of the 12D-hydroxyl of ricinoleic acid by establishing the identities and relationships of tetrols derived from each by defined chemical reactions [99]. In order to accomplish this, it was necessary to isolate four distinct diastereomeric tetrols by sequential chromatography on thin layers of silica gel impregnated with borate or arsenite. Comparable methods were utilized for resolution of diastereomers in experiments designed to determine the absolute configuration of 9,10-epoxystearic, 9,10-dihydroxystearic and 9,10,12-trihydroxystearic acids [98]. In addition, determination of the stereochemistry of the enzymic hydration of (+)-vernolic acid was facilitated by isolation of diastereomeric hydroxytosyloxyoleates by a TLC procedure [97]. Silica gel impregnated with silver nitrate was the stationary phase in this instance, and the resolution was presumed to have been helped by an interaction of the silver ions with the pi electrons of the double bond adjacent to the bulky tosyloxy group.
An alternative approach was to prepare a diastereomeric amide, i.e. of (+)-phenylethylamine, of a 2-hydroxy fatty acid for resolution by TLC or GC (see Section C.1 above) [73]. Silica gel was the adsorbent for TLC with hexane-ethyl acetate-acetic acid (70:30:2, by volume) as the mobile phase. While it was possible to obtain effective separations of the diastereomeric hydroxy fatty amides, the 2D-form migrating more rapidly, even better results were obtained when the hydroxyl group also was derivatized to the methyl ether.
The eicosanoid, (S)-5-hydroxy-6t,8c,11c,14c-eicosatetraenoic acid, was resolved from a racemic mixture after conversion to the methyl ester and then to a diastereomeric urethane derivative prepared by reaction with the isocyanate of dehydroabietylamine [38]. The separation was achieved on a preparative scale (670 mg) by low-pressure chromatography with silica gel as adsorbent and hexane-diethyl ether (5:2, v/v) as the mobile phase. The required compound was regenerated from the derivatives by appropriate hydrolysis procedures. Although the isocyanate of dehydroabietylamine is evidently a rather potent reagent for effecting resolution of diastereomeric hydroxy compounds, it is a matter for regret that it is not available from commercial suppliers.
The epimers, methyl (12S,13S,9R)- and (12S,13S,9S)-12,13-epoxy-9-hydroxy-trans-10-octadecenoate, were resolved by TLC with silica gel as adsorbent [149], confirming an earlier suggestion [47]. With compounds of this type further derivatization to facilitate resolution was not necessary.
A rather unusual procedure that has been applied to the resolution of short-chain 2-hydroxy fatty acids consisted in adding a chiral solute, i.e. copper (II) complexes of N,N-dialkyl-L-amino acids, to the mobile phase during reversed-phase HPLC [20]. It seems likely that the 2-hydroxy fatty acids form diastereomeric complexes with the chiral solute and this facilitates the separation.
Resolution of Chiral Branched-Chain Fatty Acids
An appreciable amount of work was published on the resolution of chiral isoprenoid and other multimethyl-branched fatty acids in the late 1960s and early 1970s, when capillary GC first became available, but interest in this area appears to have waned in spite of the improved opportunities from a technical standpoint now available. The topics of primary interest have been the resolution of phytanic and related acids (reviewed by Lough [85]) and of the methyl-branched acids of avian preen glands.
The pioneering work was that of Odham [101], who was able to resolve the eight diastereomers of methyl 2,4,6-trimethyloctanoate into four pairs of enantiomers by GC on a capillary column coated with polypropylene glycol; the racemates were isolated by preparative GC. With methyl 2L,4L- and 2D,4L-dimethylhexanoate, he was able to effect complete resolution by preparative GC, a separation presumably made possible by the powerful effect of the methyl substituent in position 2 [103]. Similar results were obtained with chiral trimethylnonadecanoates [104]. From the data, he was able to establish that the methyl groups in the branched-chain fatty acids from avian preen glands in a variety of species were all of the D- or (R)-configuration [101,102,105-109]. In the same way, 2D,4D,6D,8D-tetramethyloctacosanoic acid was synthesized and shown to have identical chromatographic properties to the corresponding mycocerosic acid of the tubercle bacillus [110].
Most of the important work on the resolution of the diastereomers of phytanic and related acids was carried out in the laboratory of R.G. Ackman and his collaborators. Phytanic acid, prepared chemically from phytol and consisting of the diastereomers 3(DL),7(D),11(D),15-tetramethylhexadecanoic acid, could be resolved sufficiently as the methyl ester derivatives for the relative proportions of the two epimers to be determined, the DDD-form being recognized by reference to phytanic acid isolated from Halobacterium lipids; the LDD form eluted first [3]. When the mixture of eight possible diastereomers of methyl phytanate was subjected to GC on a open-tubular column (stainless steel) coated with butanediolsuccinate polyester, three groups of partially resolved peaks were obtained [1]. Similarly, the LDD and DDD forms of methyl pristanate could be effectively resolved, but the mixture of eight possible diastereomers again gave a poorly separated triplet of peaks [3,47,91].
To improve the resolution and the confidence in the assignments, L-(-)-menthol esters of isoprenoid acids were prepared for GC analysis [2,91]. Thus with a compound such as 3(DL),7(DL),11-trimethyltridecanoic acid, the four components were distinguishable and eluted in the order LD, LL, DL and DD. While a cocktail of all eight possible diastereomers of pristanic acid eluted as a broad unresolved band, it was possible to separate a mixture of natural origin into the LDD, DLL, LLL and DDD forms as shown in Figure 9 [2,5]. Phytanic acid, as the menthyl ester, was readily separated into the LDD and DDD forms, but the all-synthetic material again gave an amorphous band [5]. Because of complex unspecified interactions between the menthyl group and the other chiral centres, it was not always possible to predict the order of elution of specific stereoisomers. The methods were applied to isoprenoid fatty acids from a variety of different sources, including fish oils and animal fats [3,5,6], bacteria [3,5,6] and oil shale [39,91]. With the exception of that from the halophilic bacterium, Halobacterium cutirubrum, which was the DDD isomer, isoprenoid fatty acids tended to be mixtures of diastereomers, the proportions of each being dependent on their origin.
Figure 9. GC separation (part chromatogram) of the (-)-menthyl ester of pristanic acid of marine origin on a stainless-steel capillary column coated with butanediolsuccinate polyester [5]. (Reproduced by kind permission of the authors and of the Journal of Chromatography, and redrawn from the original).

The potential of some of the newer chiral derivatizing agents, such as the naphthylethyl urethanes discussed above, for the resolution of diastereomeric mycocerosic acids from Mycobacteria using HPLC and TLC procedures has been briefly indicated [21].
Some Miscellaneous Separations
Short-chain 2- and 3-octanols were first successfully resolved by TLC on silica gel after conversion to diastereomeric (+)- or (-)-1-phenylethyl urethanes [45]; the mobile phase was benzene containing 5% of a polar modifier such as acetone or diethyl ether. Partial resolution of long-chain 2-alcohols in the form of diastereomeric phenylethyl or naphthylethyl urethanes was achieved by GC on packed columns with QF-1™ as the stationary phase [49]. D-Phenylpropionate derivatives of 2- and 3-alkanols [57] and drimanoyl or chrysanthemoyl derivatives of 2-octanol [31] were resolved in a similar way. Better results were obtained with the R and S forms of 2-octanol, after conversion to the diastereomeric R-(+)-2-phenylselenopropionic, alpha-methoxy-alpha-trifluoromethylphenylacetic or alpha-methoxy-alpha-methylpentafluorophenylacetic acid derivatives, by means of GC with fused-silica columns coated with SE-54™ [94]. Analogous methodology was utilized to resolve 1-octen-3-ol enantiomers [100]. In addition, HPLC and chemically bonded chiral stationary phases have been employed to resolve the 3,5-dinitrophenyl urethane derivatives of 2-alkanols [111].
Long-chain 1,2-alkanediols in the form of the bis-L-acetylmandelates were resolved by TLC on layers of silica gel with benzene-diethyl ether (97:3, v/v) as the mobile phase [18]. The method was utilized to determine the stereochemistry of these compounds in the wax esters of rat skin. Cyclic acetal and boronate derivatives of mono- and dialkyl-substituted 1,2-, 2,3-, 1,3- and 1,4-diols of low molecular weight have been resolved by GC on packed columns containing organometallic complexes with chiral substituents [123]. More recently, aliphatic 1,2-diols (C16 and C18) in the form of the 3,5-dinitrophenylurethane derivatives were resolved by HPLC with a column containing a bonded chiral phase (Sumipax™ OA-2100) [68].
D-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol, an inhibitor of glucosylceramide synthesis, was isolated from the racemic mixture by conversion to the diastereomeric R-(-)-camphanate ester for separation by HPLC on silica gel [124]. Resolution was also possible on an analytical scale by means of TLC.
Abbreviations: DNBPG, (R)-(-)-N-3,5-dinitro-benzoylphenylglycine; DNPU, 3,5-dinitrophenyl urethane; GC, gas chromatography; HPLC, high-performance liquid chromatography; TLC, thin-layer chromatography.
- Ackman, R.G., J. Chromatogr., 34, 165-173 (1968).
- Ackman, R.G., Cox, R.E., Eglinton, G., Hooper, S.N. and Maxwell, J.R., J. Chromatogr. Sci., 10, 392-401 (1972).
- Ackman, R.G. and Hansen, R.P., Lipids, 2, 357-362 (1967).
- Ackman, R.G. and Hooper, S.N., Comp. Biochem. Physiol., 24, 549-565 (1968).
- Ackman, R.G., Hooper, S.N., Kates, M., Sen Gupta, A.K., Eglinton, G. and MacLean, I., J. Chromatogr., 44, 256-261 (1969).
- Ackman, R.G., Kates, M. and Hansen, R.P., Biochim. Biophys. Acta, 176, 673-675 (1969).
- Ahnoff, M. and Einarsson, S., in Chiral Liquid Chromatography, pp. 39-80 (1989) (edited by W.J. Lough, Blackie & Son, Glasgow).
- Ando, Y. and Takagi, T., INFORM, 2, 353-354 (1991).
- Andre, J.C. and Funk, M.O., Anal. Biochem., 158, 316-321 (1986).
- Annett, R.G. and Stumpf, P.K., Anal. Biochem., 47, 638-640 (1972).
- Armstrong, D.W., Chang, C.-D. and Li, W.Y., J. Agric. Food Chem., 38, 1674-1677 (1990).
- Azerad, R., Boucher, J.L., Dansette, P. and Delaforge, M., J. Chromatogr., 498, 293-302 (1990).
- Baer, A.N., Costello, P.B. and Green, F.A., J. Lipid Res., 31, 125-130 (1990).
- Baer, A.N., Costello, P.B. and Green, F.A., Biochem. Biophys. Res. Commun., 169, 332-338 (1990).
- Baer, A.N., Costello, P.B. and Green, F.A., J. Lipid Res., 32, 341-347 (1991).
- Baer, A.N., Costello, P.B. and Green, F.A., Biochim. Biophys. Acta, 1085, 45-52 (1991).
- Baertsch, S.W., Ingram, C.D., Harris, T.M. and Brash, A.R., Biochemistry, 27, 18-24 (1988).
- Bandi, P.C. and Schmid, H.H.O., Chem. Phys. Lipids, 17, 267-274 (1976).
- Beneytout, J.L., Tixier, M. and Rigaud, M., J. Chromatogr., 351, 363-365 (1986).
- Benecke, I., J. Chromatogr., 291, 155-164 (1984).
- Besra, G.S., Minnikin, D.E. and Ridell, M., paper presented to meeting of the Royal Society of Chemistry on "Advanced Techniques in Lipid Analysis", Reading (1989).
- Bjorkhem, I. and Hamberg, M., J. Biol. Chem., 246, 7417-7420 (1971).
- Blee, E. and Schuber, F., Biochem. Biophys. Res. Commun., 173, 1354-1360 (1990).
- Borgeat, P. and Samuelsson, B., J. Biol. Chem., 254, 2643-2646 (1979).
- Borgeat, P., Hamberg, M. and Samuelsson, B., J. Biol. Chem., 251, 7816-7820 (1976).
- Brash, A.R., Baertsch, S.W., Ingram, C.D. and Harris, T.M., J. Biol. Chem., 262, 15829-15839 (1987).
- Brash, A.R. and Hawkins, D.J., Methods Enzymol., 187, 187-195 (1990).
- Brash,A .R., Ingram, C.D. and Harris, T.M., Biochemistry, 26, 5465-5471 (1987).
- Brash, A.R., Porter, A.T. and Maas, R.L., J. Biol. Chem., 260, 4210-4216 (1985).
- Breckenridge, W.C. in Handbook of Lipid Research. Vol. 1, pp. 197-232 (1978) (edited by A. Kuksis, Plenum Press, New York).
- Brooks, C.J.W., Gilbert, M.T. and Gilbert, J.D. Anal. Chem., 45, 896-902 (1973).
- Camacho, P.L., Geiger, E., Vigh, G., Webster, R. and Thompson, D.H., J. Chromatogr., 506, 611-616 (1990).
- Christie, W.W., in Analysis of Oils and Fats, pp. 313-319 (1986) (edited by R.J. Hamilton and J.B. Rossell, Elsevier Applied Science, London).
- Christie, W.W., High-Performance Liquid Chromatography and Lipids (1987) (Pergamon Press, Oxford).
- Christie, W.W., Gas Chromatography and Lipids (1989) (The Oily Press, Ayr).
- Christie, W.W., Nikolova-Damyanova ,B., Laakso, P. and Herslof, B., J. Am. Oil Chem. Soc., 68, 695-701 (1991).
- Claeys, M., Coene, M.-C., Herman, A.G., Jouvenaz, G.H. and Nugteren, D.H., Biochim. Biophys. Acta, 713, 160-169 (1982).
- Corey, E.J. and Hashimoto, S., Tetrahedron Lett., 22, 299-302 (1981).
- Cox, R.E., Maxwell, J.R., Eglinton, G., Pillinger, C.T., Ackman, R.G. and Hooper, S.N., Chem. Commun., 1639-1641 (1970).
- Engel, K.-H., Flath, R.A., Albrecht, W. and Tressl, R., J. Chromatogr., 479, 176-180 (1989).
- Engel, K.-H., Albrecht, W. and Heidlas, J., J. Agric. Food Chem., 38, 244-247 (1990).
- Fahlstadius, P. and Hamberg, M., Chem. Phys. Lipids, 51, 15-22 (1989).
- Falck, J.R., Manna, S., Jacobson, H.R., Estabrook, R.W., Chacos, N. and Capdevila, J., J. Am. Chem. Soc., 106, 3334-3336 (1984).
- Falgueyret, J.-P., Leblanc, Y., Rokach, J. and Riendeau, D., Biochem. Biophys. Res. Commun., 156, 1083-1089 (1988).
- Freytag, W. and Ney, K.H., J. Chromatogr., 41, 473-474 (1969).
- Gardner, H.W., Biochim. Biophys. Acta, 1001, 274-281 (1989).
- Gardner, H.W. and Kleiman, R., Biochim. Biophys. Acta, b, 113-125 (1981).
- German, J.B. and Berger, R., Lipids, 25, 849-853 (1990).
- Hamberg, M., Chem. Phys. Lipids, 6, 152-158 (1971).
- Hamberg, M., Anal. Biochem., 43, 515-526 (1971).
- Hamberg, M., Biochim. Biophys. Acta, b, 191-197 (1983).
- Hamberg, M., Lipids, 26, 407-415 (1991).
- Hamberg, M., J. Agric. Food Chem., 39, 1568-1572 (1991).
- Hamberg, M. and Samuelsson, B., Proc. Nat. Acad. Sci. USA, b, 3400-3404 (1974).
- Hammarstrom,S ., FEBS Lett., 5, 192-195 (1969).
- Hammarstrom, S., Methods Enzymol., 35, 326-334 (1975).
- Hammarstrom, S. and Hamberg, M., Anal. Biochem., 52, 169-179 (1973).
- Hammonds, T.D., Blair, I.A., Falck, J.R. and Capdevila, J.H., Anal. Biochem., 182, 300-303 (1989).
- Hawkins, D.J. and Brash, A.R., J. Biol. Chem., 262, 7629-7634 (1987).
- Hawkins, D.J., Kuhn, H., Petty, E.H. and Brash, A.R., Anal. Biochem., 173, 456-462 (1988).
- Hitchcock, C. and Rose, A., Biochem. J., 125, 1155-1156 (1971).
- Hughes, M.A. and Brash, A.R., Biochim. Biophys. Acta, 1081, 347-354 (1991).
- Hurst, J.S., Balazy, M., Bazan, H.E.P. and Bazan, N.G., J. Biol. Chem., 266, 6726-6730 (1991).
- Itabashi, Y., Kuksis, A., Marai, L. and Takagi, T., J. Lipid Res., 31, 1711-1717 (1990).
- Itabashi, Y., Kuksis, A. and Myher, J.J., J. Lipid Res., 31, 2119-2126 (1990).
- Itabashi, Y. and Takagi, T., Lipids, 21, 413-416 (1986).
- Itabashi, Y. and Takagi, T., J. Chromatogr., 402, (1987).
- Itabashi, Y., Takagi, T. and Tsuda,T., J. Chromatogr., 472, 271-276 (1989).
- IUPAC-IUB Commission on Biochemical Nomenclature., Eur. J. Biochem., 2, 127-131 (1967); Biochem. J., 105, 897-902 (1967).
- IUPAC-IUB Commission on Biochemical Nomenclature., Hoppe-Seyler's Z. Physiol. Chem., 358, 599-616 (1977); J. Lipid Res., 19, 114-125 (1978).
- Kagan, H., Organic Stereochemistry (1979) (Edward Arnold, London).
- Karara, A., Dishman, E., Falck, J.R. and Capdevila, J.H., J. Biol. Chem., 266, 7561-7569 (1991).
- Karlsson, K.-A. and Pascher, I., Chem. Phys. Lipids, 12, 65-74 (1974).
- Kitamura, S., Shimizu, T., Miki, I., Izumi, T., Kasama, T., Sato, A., Sano, H. and Seyama, Y., Eur. J. Biochem., 176, 7225-7231 (1988).
- Kodali, D.R., J. Lipid Res., 28, 464-469 (1987).
- Konig, W.A., Lutz ,S., Colberg, C., Schmidt, N., Wenz, G., von der Bey, E., Mosandl, A., Gunther, C. and Kustermann, A., J. High Resolut. Chromatogr., Chromatogr. Commun., 11, 621-625 (1988).
- Kuhn, H., Belkner, J. and Wiesner, R., Eur. J. Biochem., 191, 221-227 (1990).
- Kuhn, H., Heydeck, D. and Sprecher, H., Biochim. Biophys. Acta, 1081, 129-134 (1991).
- Kuhn, H., Sprecher, H. and Brash, A.R., J. Biol. Chem., 265, 16300-16305 (1990).
- Kuhn, H. and Wiesner, R., J. Chromatogr., 520, 391-401 (1990).
- Kuhn, H., Wiesner, R., Alder, L. and Schewe, T., Eur. J. Biochem., 186, 155-162 (1989).
- Kuhn, H., Wiesner,R ., Lankin, V.Z., Nekrasov, A., Alder, L. and Schewe, T., Anal. Biochem., 160, 24-34 (1987).
- Kuksis, A., Marai, L., Myher, J.J. and Itabashi, Y., in Proceedings 15th Scandinavian Symposium on Lipids, Rebild Bakker, Denmark, pp. 336-370 (1989) (edited by V.K.S. Shukla and G. Holmer, Lipidforum).
- Laakso, P. and Christie, W.W., Lipids, 25, 349-353 (1990).
- Lough, A.K., Prog. Lipid Res., 14, 5-48 (1975).
- Lough, W.J. (Editor), Chiral Liquid Chromatography (1989) (Blackie & Son, Glasgow).
- Lysz, T.W., Wu, Y., Brash, A., Keeting, P.E., Lin, C. and Fu, S.C.J., Current Eye Res., 10, 331-337 (1991).
- Maas, R.L., Brash, A.R. and Oates, J.A., Proc. Natl. Acad. Sci. USA, 78, 5523-5527 (1981).
- Maas, R.L., Ingram, C.D., Porter, A.J., Oates, J.A., Taber, D.F. and Brash, A.R., J. Biol. Chem., 260, 4217-4228 (1985).
- Maas, R.L., Turk, J., Oates, J.A. and Brash, A.R., J. Biol. Chem., 257, 7056-7067 (1982).
- MacLean, I., Eglinton, G., Douraghi-Zadeh, K., Ackman, R.G. and Hooper, S.N., Nature, 218, 1019-1024 (1968).
- Meijer, L., Brash, A.R., Bryant, R.W., Ng, K., Maclouf, J. and Sprecher, H., J. Biol. Chem., 261, 17040-17047 (1986).
- Michelsen, P., Aronsson, E., Odham, G. and Akesson, B., J. Chromatogr., 350, 417-426 (1985).
- Michelsen, P. and Odham, G., J. Chromatogr., 331, 295-302 (1985).
- Miller, L. and Bush, H., J. Chromatogr., 484, 337-345 (1989).
- Miller, L. and Weyker, C., J. Chromatogr., 511, 97-107 (1990).
- Morris, L.J. and Crouchman, M.L., Lipids, 4, 50-54 (1969).
- Morris, L.J. and Crouchman, M.L., Lipids, 7, 372-379 (1972).
- Morris, L.J. and Wharry, D.M., Lipids, 1, 41-46 (1966).
- Mosandl, A., Heusinger, G. and Gessner, M., J. Agric. Food Chem., 34, 119-122 (1986).
- Odham, G., Ark. Kemi, 23, 431-451 (1965).
- Odham, G., Ark. Kemi, 25, 543-554 (1966).
- Odham, G., Ark. Kemi, 26, 367-377 (1966).
- Odham, G., Ark. Kemi, 27, 231-250 (1967).
- Odham, G., Ark. Kemi, 27, 251-255 (1967).
- Odham, G., Ark. Kemi, 27, 263-288 (1967).
- Odham, G., Ark. Kemi, 27, 289-284 (1967).
- Odham, G., Fette Seifen Anstrichm., 69, 164-172 (1967).
- Odham, G. and Stenhagen, E., Acc. Chem. Res., 4, 121-128 (1971).
- Odham, G., Stenhagen, E. and Waern, K., Ark. Kemi, 31, 533-554 (1970).
- Oi, N. and Kitahara, H., J. Chromatogr., 265, 117-120 (1983).
- Oliw, E.H., J. Chromatogr., 526, 525-529 (1990).
- Oliw, E.H. and Sprecher, H., Biochim. Biophys. Acta, 1002, 283-291 (1989).
- Porter, N.A., Dussault, P., Breyer, R.A., Kaplan, J. and Morelli, J., Chem. Res. Toxicol., 3, 236-243 (1990).
- Pirkle, W.H. and Adams, P.E., J. Org. Chem., 45, 4111-4117 (1980).
- Pirkle, W.H. and Hauske, J.R., J. Org. Chem., 42, 1839-1844 (1977).
- Pirkle, W.H. and Hauske, J .R., J. Org. Chem., 42, 2781-2782 (1977).
- Pirkle, W.H. and Hoekstra, M.S., J. Org. Chem., 39, 3904-3906 (1974).
- Pirkle, W.H. and House, D.W., J. Org. Chem., 44, 1957-1960 (1979).
- Rackham, D.M. and Harvey, G.A., J. Chromatogr., 542, 189-192 (1991).
- Ramsey, O.B., Stereochemistry: Nobel Prize Topics in Chemistry (1981) (Heyden, London).
- Rogalska, E., Ransac, S. and Verger, R., J. Biol. Chem., 265, 20271-20276 (1990).
- Schurig, V. and Wistuba, D., Tetrahedron Lett., 25, 5633-5636 (1984).
- Shukla, A. and Radin, N.S., J. Lipid Res., 32, 713-722 (1991).
- Smith, C.R., in Topics in Lipid Chemistry. Vol. 1, pp. 277-368 (1970) (edited by F.D. Gunstone, Logos Press, London).
- Smith, C.R., in Topics in Lipid Chemistry. Vol. 3, pp. 89-124 (1972) (edited by F.D. Gunstone, Paul Elek (Scientific Books), London).
- Smith, C.R., J. Chromatogr. Sci., 14, 36-39 (1976).
- Sonnet, P.E., Piotrowski, E.G. and Boswell, R.T., J. Chromatogr., 436, 205-217 (1988).
- Takagi, T., Prog. Lipid Res., 29, 277-298 (1990).
- Takagi, T. and Ando, Y., Lipids, 25, 398-400 (1990).
- Takagi, T. and Ando,Y ., J. Japan Oil Chem. Soc., 39, 622-628 (1990).
- Takagi, T. and Ando, Y., INFORM, 2, 353 (1991).
- Takagi, T. and Ando, Y., Lipids, 26, 542-547 (1991).
- Takagi, T. and Ando, Y., J. Japan Oil Chem. Soc., 40, 288-292 (1991).
- Takagi, T. and Itabashi, Y., Yukagaku, 34, 962-963 (1985).
- Takagi, T. and Itabashi, Y., J. Chromatogr., 366, 451-455 (1986).
- Takagi, T. and Itabashi, Y., Lipids, 22, 596-600 (1987).
- Takagi, T. and Itabashi, Y., J. Chromatogr. Sci., 27, 574-577 (1989).
- Takagi, T., Okamoto, J., Ando, Y. and Itabashi, Y., Lipids, 25, 108-110 (1990).
- Takagi, T. and Suzuki, T., J. Chromatogr., 519, 237-243 (1990).
- Takahashi, Y., Ueda, N. and Yamamoto, S., Arch. Biochem. Biophys., 266, 613-621 (1988).
- Tatsumi, K., Kishimoto, Y. and Hignite, C., Arch. Biochem. Biophys., 165, 656-664 (1974).
- Tsai, S.-C., Steinberg, D., Avigan, J. and Fales, H.M., J. Biol. Chem., 248, 1091-1097 (1973).
- Turk, J., Stump, W.T., Wolf, B.A., Easom, R.A. and McDaniel, M.L., Anal. Biochem., 174, 580-588 (1988).
- Turk, J., Wolf, B.A., Easom, R.A., Hughes, J.H. and McDaniel, M.L., Biochim. Biophys. Acta, 1001, 16-24 (1989).
- van der Donk, E.M.M., Verhagen, J., Veldink, G.A. and Vliegenthart, J.F.G. Biochim. Biophys. Acta, 1081, 135-140 (1991).
- Van Os, C.P.A., Rijke-Schilder, G.P.M., Kamerling,J .P., Gerwig, G.J. and Vliegenthart, J.F.G., Biochim. Biophys. Acta, 620, 326-331 (1980).
- Van Os, C.P.A., Rijke-Schilder, G.P.M., Van Halbeek, H., Verhagen, J. and Vliegenthart, J.F.G., Biochim. Biophys. Acta, 663, 177-193 (1981).
- Van Os, C.P.A., Vliegenthart, J.F.G., Crawford, C.G. and Gardner, H.W., Biochim. Biophys. Acta, 713, 173-176 (1982).
- Weaner, L.E. and Hoerr, D.C., Anal. Biochem., 160, 316-322 (1987).
- Weaner, L.E. and Hoerr, D.C., J. Chromatogr., 437, 109-119 (1988).
- Westley, J.W. and Halpern, B., J. Org. Chem., 33, 3978-3980 (1968).
- Westlund, P., Edenius, C. and Lindgren, J.A., Biochim. Biophys. Acta, 962, 105-115 (1988).
- Westlund, P., Palmblad, J., Falck, J.R. and Lumin, S., Biochim. Biophys. Acta, 1081, 301-307 (1991).
- Wiesner, R. and Kuhn, H., Suppl. Chromatographie, 3, 32-38 (1989).
- Wiesner, R. and Kuhn, H., Proc. Int. Symp. Instrumentalized Anal. Chem. Comp. Technol., 448-457 (1990).
- Wood, R., Bever, E.L. and Snyder, F., Lipids, 1, 399-408 (1966).
- Woollard, P.M., Biochem. Biophys. Res. Commun., 136, 169-176 (1986).
- Yang, L.-Y. and Kuksis, A., J. Lipid Res., 32, 1173-1186 (1991).
- Zief, M. and Crane, L.J. (Editors), Chromatographic Chiral Separations. Chromatographic Science Series. Vol. 40 (1988) (Marcel Dekker, New York).
In This Section
- Solid-phase extraction columns in the analysis of lipids
- Preparation of Ester Derivatives of Fatty Acids for Chromatographic Analysis
- Preparation of Lipid Extracts Tissues
- The Chromatographic Resolution of Chiral Lipids
- Detectors for HPLC of Lipids with Special Reference to Evaporative Lght-Scattering Detection
- Why Doesn't Your Method Work When I Try It?
- Laboratory Accreditation in a Lipid Analysis Context
- What Column do I Need for Gas Chromatographic Analysis of Fatty Acids?
- Fatty Acid Analysis by HPLC
- Alternatives to Methyl Esters for GC Analysis of Fatty Acids
- A Practical Guide to the Analysis of Conjugated Linoleic Acid (CLA)
- Application of Infrared Spectroscopy to the Rapid Determination of Total Saturated, trans, Monounsaturated, and Polyunsaturated Fatty Acids
- The Use of Lithiated Adducts for Structural Analysis of Acylglycerols by Mass Spectrometry with Electrospray Ionization
- Identification of FAME Double Bond Location by Covalent Adduct Chemical Ionization (CACI) Tandem Mass Spectrometry
- The Use of Countercurrent Chromatography (CCC) in Lipid Analysis
- Gas Chromatographic Analysis of Plant Sterols
- Analysis of Tocopherols and Tocotrienols by HPLC
- Reversed-Phase HPLC of Triacylglycerols
- Structural Analysis of Triacylglycerols
- Thin-Layer Chromatography of Lipids
- High-temperature Gas Chromatography of Triacylglycerols
- Modification of an AOCS Official Method for Crude Oil Content in Distillers Grains and Other Agricultural Materials
- Lipidomics - A Personal View