Characterization of Lipids by MALDI Mass Spectrometry
The Author: Jackson O. Lay, Jr., Director, Arkansas Statewide Mass Spectrometry Facility, University of Arkansas
Introduction
Because of their low volatility, lipids were until recently analysed mainly via their component parts, especially by gas chromatography. However, direct analysis of lipids without resort to chemical treatment and derivatization is simpler and faster. The development of desorption ionization techniques such as matrix assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) have given mass spectrometry users tools to perform experiments on non-volatile compounds. MALDI involves ablation of the analyte from a planar surface, usually co-deposited with a large excess of a solid matrix. MALDI is typically not coupled with on-line separation technique, but is well suited to implementation with sensitive, simple, and fast-scanning time-of-flight (TOF) mass analyzers. ESI, on the other hand, can be implemented on a variety of mass spectrometers and is based on introduction of a liquid, usually in a protic solvent, often directly coupled to a reversed phase HPLC. While both ESI and MALDI can be used for the direct analysis of lipids, this report focuses only on MALDI and in particular with respect to the analysis of triacylglycerols and bacterial phospholipids.
Theory
Sample Preparation and MALDI Ionization: The ion source consists of a pulsed laser focused to a precise location on a planar, electrically conducting (usually metal) target plate. Often many samples are deposited in known locations in the x,y plane, allowing multiple samples to be accessed as needed without breaking vacuum. Usually the system includes a video display with sufficient magnification to see individual crystals of the matrix in which the sample is deposited.
The matrix is a low volatility substance that absorbs light at the fixed wavelength of the pulsed laser. This indirectly couples the energy form the laser with any analyte embedded within the matrix, allowing its’ non-equilibrium desorption from the surface to the vapor state without the heating that would be normally be associated with an equilibrium-based process. A novel aspect of MALDI with lipids is the amount of matrix used. While it is typical in the analysis of proteins to use a massive excess (> 1000:1 w/w or v/v) compared to analyte, for lipids MALDI is best accomplished when the amount of lipid is more comparable to the amount of matrix. In fact there have been some reports on the characterization of triacylglycerols (TAGs) by direct laser desorption, without any matrix. The amount of matrix is typically adjusted by trial and error to produce the best lipid mass spectra.
Because the matrix is typically a weak acid lipids can be ionized by electrostatic association with a cation before, during or after desorption, giving rise to gas-phase ions. The usual charge carrying species is a proton, giving rise to an “[M+H]+” ion, 1 Dalton higher in mass than the neutral precursor. With their significant affinity for alkali metals, the lipids may also desorb from the matrix as sodium or potassium adducts ([M+X]+, X = Na or K). Even when these alkali cations are not deliberately added before measurements, they are frequently encountered because of their ubiquitous nature. While it is possible that the lipids might desorb with more than two charge carrying species (z > 1) and appear at a fraction of the expected mass (MS measures m/z) this is a low probability event for MALDI and most ions are singly charged.
Fragmentation of Lipid Ions: Fragmentation in MALDI is not normally a major process, and MALDI is often referred to as a “soft ionization” technique. Typically the analyte is represented largely, if not entirely, by an intense singly cationized species. However for lipids, the spectra can be significantly different than might be expected. The stability of cationized lipids is dependent upon both the class of the lipid and the charge carrying species. For triacylglycerols (TAGs), protonation gives rise to a short lived species that might not be observed in the mass spectrum because it fragments promptly [1]. For this reason the spectra might show enhanced (or only) sodium adduct ions in the m/z region where the cationized species would be expected, even if no alkali cations are added to the sample.
Just as important is the observation of the measurement induced fragments. Prompt fragmentation of protonated TAGs in the MS ion source gives rise to measurement-induced “diacylglycerol (DAG)-like” ions. The net effect is an exaggeration of the extent of alkali cation content and the apparent presence of DAGs. While sodium and potassium adduct ions in MALDI mass spectra are not at all unusual, the observation of the apparent DAGs can cause significant confusion in lipids analysis. The magnitude of this artifact in the MALDI process can be discerned for any given set of experimental conditions simply by analysis of food-grade TAG known not to contain DAGs. The intensity of these DAGs artifacts can be comparable to the stable sodium (or potassium) adducts with the parent TAGs. This artifact can be reduced or entirely eliminated by modification of the matrix to prevent formation of the protonated molecules which dissociate to give these artifacts. Addition of sodium hydroxide to the matrix will result in formation of its sodium salt as protons are depleted by combination with hydroxide ions to form water. In this situation, the predominant ionic form of the analyte is as the stable sodium adduct. While this minimally affects the signals for the sodium adducts of the TAG already observed before addition of base to the matrix, it does reduce or eliminate the production of the “measurement-induced” DAGs ions. By eliminating measurement induced DAGs it then becomes possible to measure any “real” DAGs in the sample without the complication of additional measurement-induced species.
Selective Ionization: Because the ionization process is based on acid-base chemistry, the relative abundance of ions in spectra is not always a good reflection of the actual relative abundances in a mixture. For example, the MALDI-friendly phosphatidylcholine, if present in any lipid mixture, will generally suppress other lipids and thus generally appear over-represented based on ion intensities. As the difference in polarity between it and the other lipids in a mixture increases, the magnitude of the suppression of the other components increases as well. Nevertheless, the relative signal strength can be a good representation of the approximate amounts for pure compounds or for mixtures when all of the components have comparable chemical and physical properties.
Because the constituent TAGs are chemically similar, food oils can be analyzed to obtain a “fingerprint” with relative abundances of the desorbed/ionized TAGs that reasonably reflect their relative abundances in the mixture [2]. Likewise, crude lipid extracts from bacteria, which contain primarily phospholipids, can also give a good approximation of the overall lipid profile for taxonomic identification [3]. In more complex mixtures the polar phospholipids normally swamp the signals from less polar TAGs. In the applications described above, this suppression had a minimal impact because the mixtures did not contain significant amounts of both polar and non-polar lipids. On the other hand, the characterization of the non-polar lipids in a mixture also containing polar lipids, such as a tissue extract, direct MALDI is not practical. Direct MALDI on total ground beef lipids extracts typically misses the abundant TAGs and detects only the less abundant but more polar lipids. The simplest approach in this case would be to fractionate the samples by solid phase extraction (SPE) into a polar and a non-polar fraction and analyze each separately by direct MALDI [4]. Alternative approaches are collection of HPLC fractions or direct analysis of TLC spots. MALDI has been applied directly to TLC plates to obtain mass spectra of spatially separated lipids [5].
Interpretation of MALDI Mass Spectra of Lipids
Triacylglycerols: To a first approximation, the MALDI mass spectrum will include sodium adduct ions, (M+23), for each of the TAGs present in the food oil. They will all appear in the “TAGs region” of the spectrum. The boundaries of this mass region can be calculated based on the expected fatty acid composition. The mass of the glycerol backbone contributes 89 Daltons. The three fatty acids each contribute their free acid mass less water. The mass of a TAG containing three C16:0 fatty acids would be 806 Daltons. The charge carrying sodium cation forming the sodium-TAG adduct would be observed 23 Daltons higher in mass at 829 Daltons. For each substitution of a C16 fatty acid with a C18 fatty acid, the mass is increased by 28 Daltons for the same degree of unsaturation. Thus a TAG containing only saturated C18 fatty acids would be observed as a sodium adduct ion at m/z 913. Likewise ions near m/z 857 and 885 would then have two C16/one C18 and one C16/two C18 fatty acids respectively. The expected mass region for the intact TAGs ions (as sodium adducts) for typical plant-based food oils (C16 /C18 fatty acids) would be between about 700 and 1000 Daltons (Table 1).
| Table 1. Mass and likely composition (from most abundant fatty acids) for common edible vegetable oil TAGs. | |||||
| Nominal Mass1 | TAG2 | Fatty Acids3 | Nominal Mass1 | TAG2 | Fatty Acids3,4 |
|---|---|---|---|---|---|
| 829 | C48:0 | PPP | 883 | C52:1 | PSO |
| 851 | C50:3 | PPLn | 885 | C52:0 | PSS |
| 853 | C50:2 | PPL | 899 | C54:7 | LLLn |
| 855 | C50:1 | PPO | 901 | C54:6 | LLL |
| 857 | C50:0 | PPS | 903 | C54:5 | OLL3,4 |
| 873 | C52:6 | PLnLn | 905 | C54:4 | OOL |
| 875 | C52:5 | PLLn | 907 | C54:3 | OOO |
| 877 | C52:4 | PLL3,4 | 909 | C54:2 | SOO |
| 879 | C52:3 | POL | 911 | C54:1 | SSO |
| 881 | C52:2 | POO | 931 | C54:0 | SSS |
| 1 sodium adduct ion 2 xx:yy carbon number:double bonds |
3 P =16:0, S = 18:0, O = 18:1, L = 18:2, Ln = 18:3 4 except canola or flaxseed |
||||
Phospholipids: The mass spectra of phospholipids are more complicated than those of the TAGs. These lipids may show both protonated molecules and alkali-metal adducts. They may also show fragment ions ranging from loss of a fatty acid moiety to an ion corresponding to the polar head group. In a mixture it is often necessary to use MS/MS experiments to sort out which fragments are most likely to be associated with specific parent ions. In some cases, the parent ions may not be observed. Finally, the highly polar nature of the phospholipids results in significant suppression of other polar lipids and almost complete suppression of non-polar lipids. In general the mass spectra of phospholipid mixtures should be interpreted with more caution than those of TAGs and often based on analysis of reference materials or very high mass accuracy .
Edible Oils (TAGs)
Characterization of edible oils is of considerable interest because of the economic fraud arising from price differences amongst types and qualities of oils. The classical approach of GC (or GC/MS) of fatty acid methyl esters (FAMEs)has been the method of choice for characterization of food oils, although approaches have recently been developed based on HPLC and atmospheric pressure chemical ionization (APCI) MS. Both of these approaches require chromatography and FAMEs also involves cleavage/derivatization. Direct analysis without derivatization or chromatography is a faster approach. MALDI can be used to characterize food oils based on the characteristic ratios of TAGs. For many oils, typically have major differences in fatty acid compositions, the general mass spectral profile can be easily used to distinguish one type from another by inspection. Using this approach and some knowledge of the predominate fatty acids in foods, it is possible to obtain semi-quantitative information about the relative amounts of the fatty acids present. If the intensity of the TAG peaks is partitioned amongst the expected fatty acids (see below) and the values are summed, the relative fractions of the total correlate well with the known relative compositions of the FAs.
Figure 1 shows the TAGs region MALDI mass spectrum of a commercial soybean oil sample. One cluster of ions near m/z 901corresponds to TAGs having three C18 fatty acids and either two (m/z 909), three (m/z 907), four (m/z 905), five (m/z 903), six (m/z 901) or seven (m/z 899) double bonds shared amongst the three fatty acids. The ions approximately 22 Daltons lower in mass are not the corresponding protonated molecules, despite the correspondence in mass. They are ions from TAGs that include a C16:0 fatty acid. Because this substation involves exchange of a partially unsaturated C18 fatty acid (mostly C18:2) with a C16:0, the mass shift for the cluster is 24 Daltons rather than the expected 28 Daltons (28 if the degree of saturation had remained the same). The carbon isotopes in the spectra give an additional peak one Dalton higher in mass, whereas the mass differences of 2 Daltons correspond to a degree of unsaturation.
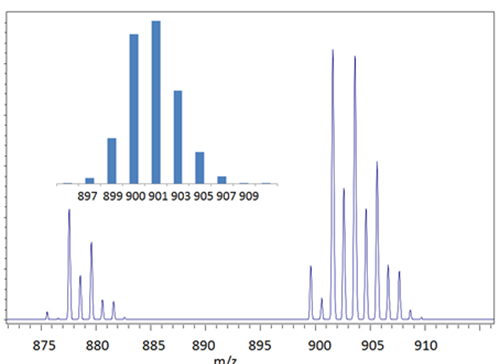
Figure 1. The TAGs region of the MALDI mass spectra obtained from a soybean sample and a theoretical profile (inset) for the (C18)3 TAGs.
A typical distribution of the major fatty acids in soy oil is as follows: C18:3 (7%), C18:2 (54%), C18:1 (24%), C18:0 (4%) and C16:0 (11%). Using these values, an expected composition for the C18 TAGs is presented as an inset in Figure 1 and in Table 2. The expected low abundance TAGs at m/z 895, 897, 911 and 913 are not observed. Two C52 TAGs (C18+C18+C16:0) are also expected in the spectrum at m/z 877 and 879 with approximately 46 and 37% relative abundance. While the agreement between calculated and expected peak intensities is not perfect, the values are sufficiently close that approximate fatty acid compositions can be calculated starting from the TAG intensities. Consider for example evaluation of the spectrum in Figure 1 as an unknown. Interpretation can be simplified because of the following characteristics of food-related TAGs. At the fatty acid level, the carbon and double bond numbers can be associated primarily with one fatty acid. For example C10:0, C12:0, C14:0, C16:0, C18:0, C18:1, C18:2 and C18:3 are primarily capric, lauric, myristic, palmitic (P), steric (S), oleic (O), linoleic (L), and linolenic (Ln) acids, respectively. The distribution of these fatty acids in biological systems is not random. All possible chemical and fatty acid combinations are not found. Carbon numbers are usually even (see an interesting exception below for Escherichia coli.). In different classes of oils, certain fatty acids predominate. In nature, the major C10, C12, C14, and C16 fatty acids are usually saturated.
| Table 2. Assignment of TAGs in the MALDI mass spectrum of a soybean oil sample | |||||
| Nominal Mass1 | Relative Abundance [Observed (Expected) %] | Primary TAG Fatty Acids (Alternative Compositions2) | Nominal Mass1 | Relative Abundance [Observed (Expected) %] | Primary TAG Fatty Acids (Alternative Compositions2) |
|---|---|---|---|---|---|
| 873 | 0 (1) | LnLnP | 899 | 20 (28) | LLLn |
| 875 | 3 (11) | PLLn | 901 | 100 (92) | LLL (LLnO) |
| 877 | 42 (46) | PLL | 903 | 98 (100) | OLL |
| 879 | 29 (37) | PLO | 905 | 58 (57) | OOL (LLS) |
| 881 | 7 (8) | POO | 907 | 18 (20) | OOO (LOS) |
| 883 | 0 (3) | PSO | 909 | 1 (4) | SOO (LSS) |
| 885 | 0 (0.2) | PSS | 911 | 0 (1) | OSS |
| 895 | 0 (0.2) | LnLnLn | 913 | 0 (0.03) | SSS |
| 897 | 0 (3) | LnLnL | |||
| 1 sodium adduct ions 2 >minor TAGs with >10% the abundance of the main composition. |
|||||
These fatty acids can often be assigned their contribution to the total TAGs FAs based on the total carbon number for the TAG. Thus, in plant oils a C52 TAG is most likely comprised of a C16 and two C18 fatty acids. A C16 fatty acid is almost certainly C16:0. Any double bonds must be shared between the remaining two C18 FAs. If there were three double bonds and 36 carbons, for many common vegetable oils C18:1 and C18:2 are much more likely than C18:0 and C18:3. Flaxseed oil does have a very high abundance of C18:3, but like olive oil (mostly C18:1), it gives a unique mass spectral fingerprint (data not shown) that is easily recognized. Shea nut and cocoa butter have a significant C18:0, also giving diagnostic ions in the TAGs mass spectrum, and likewise these oils can also be recognized by inspection. Non-vegetable oils like lard and tallow also have unique characteristics. Both have a more significant contribution from C16:0 and also some C14:0. The butter fats have significant C4-C12 fatty acids
With a little biological information, the most likely FA composition of TAGs can be deduced, and then the total intensity partitioned appropriately. The masses of the TAGs ions associated with specific expected fatty acid combinations for typical food related vegetable oils are listed in Table 1. Alternative tables can be developed for animal fats, unusual plants or other general sources of lipids and used the same way. It should be noted that assignment of masses to a most likely FA composition is possible because of both the biology and the instability of the protonated TAGs. If these TAGs formed both proton and sodium adduct ions, there would be many isobaric masses. When estimating the total FA composition, the alternative compositions for a given mass are simply assumed not to be present, only the values in the table are used. The TAG masses and intensities observed in the soybean oil mass spectrum in Figure 1 are listed in Table 2 alongside the proposed composition (last column). To give a basis for comparison, the expected ion intensities for the TAGs were also calculated using a typical soy oil fatty acid composition. It should be noted that these expected values are from averages, they are not for this specific sample, and they should only be considered a guide. As noted above, the lower abundance TAGs were not observed. Nevertheless, the intensity values for the TAGs that are observed using this approach generally fall reasonably close to expected values. For this reason it is possible to work backwards and use the TAGs intensities to calculate the FA composition. Summing up the intensity contribution for each of the fatty acids assigning them based on the expected FAs in each TAG (ignoring minor contributions) and renormalizing the values gives the approximate fatty acid composition shown in Table 3. For this soybean oil sample the value for Ln (2%) was lower than the expected range (5-9%) whereas the value for L (63%) was higher than expected (48-58%). The values for O (28%) and P (7%) were within the expected range. The expected small contribution of S (2-6%) was not detected.
Fatty AcidEstimated Value (%)Accepted Values1 Soybean (%)Accepted Values1 Olive (%)Accepted Values1 Corn (%)
| Table 3. Estimated fatty acid composition of a commercial soybean oil sample by direct MALDI TOF, the accepted range of values, and a comparison with accepted values for two other high volume commodity food oils. | ||||
| Ln | 2 | 5-9 | <2 | <2 |
| L | 63 | 48-58 | 4-15 | 34-65 |
| O | 28 | 19-30 | 65-85 | 19-50 |
| S | 0 | 2-6 | 1-3 | 0.5-4 |
| P | 7 | 7-12 | 7-16 | 8-19 |
| 1 Values for products listed by Welch, Holme and Clark Co. Inc. | ||||
This approach gives a semi-quantitative estimate of FA composition. However, with this level of uncertainty, and with all of the underlying simplifications, this rapid MALDI method would not differentiate this oil sample from a corn oil sample using the wide range of literature values, but it could easily differentiate it from an olive oil (Table 3) and many other oils (data not shown) using just the range of values reported in the literature. In practice the results obtained using co-analysis of reference samples rather than the full range of literature values provides a much better match. Issues related to suppression of minor components in the mixture or dynamic range issues as well as differences between methods are more easily accounted for by comparison of authentic samples run on the same instrument and using the same approach. In side by side comparisons, in most cases it is possible to differentiate soy and corn oil by direct MALDI using analysis of reference samples run at the same time and on the same instrument [2].
Bacterial Lipids (Phospholipids)
There has always been a need for reliable methods for the rapid identification of bacteria to facilitate the timely treatment of human disease. Disease and death associated with unidentified pathogens is an ongoing problem even in developed countries. Considering only food borne illness, in the United States the CDC reports a significant number of deaths and illness each year. In many cases the causative microorganism is not known. Clearly additional methods are needed to complement existing approaches for rapid identification [7]. The most common mass spectral method developed for taxonomic identification of bacteria is based on the FAMEs produced from bacterial lipids. Comprehensive libraries have been developed that allow bacteria that can be cultured to be identified based on matching profiles. This approach provides reliable identification to the genus, species, and even the strain level.
Direct analysis of the bacterial lipids represents an alternative and potentially much faster approach. Simple approaches for recovering lipids from biological samples were developed by Folch et al. [8] and Bligh and Dyer [9]. Slightly safer solvents and much smaller volumes can be substituted. For example, rather than using chloroform and methanol, we substitute dichloromethane and ethanol. Using one approach, bacterial cells are mixed with 30 mL of a 1:1:1 (v:v:v) mixture of these solvents and water. The mixture is shaken briefly and allowed to stand for a few hours or overnight. After phase separation, the bottom organic layer, containing the lipids, is recovered and concentrated to 1 mL. Because of the volatility of dichloromethane this can be accomplished easily using a stream of nitrogen. A single microlitre of this lipid extract is sufficient to produce taxonomically useful spectra. This approach can be scaled up or down depending on the amount of material available. In some cases the organic solution can be analyzed directly without concentration.
Figure 2 shows part of the MALDI mass spectrum (from m/z 700-800) of the organic extract from E. Coli. For C16 to C18 fatty acids, the diacylglycerols and phospholipids would be expected primarily in the range of 600-800 Da, whereas triacylglycerols with the same fatty acids would be expected around 800-1000 Da. Because of their much higher polarity, it is expected that any phospholipids would suppress whatever triacylglycerols might be present, and no signals are seen in the TAGs region (data not show). In this spectrum the ions are attributed to the phospholipid phosphatidylethanolamine (PE) species having a total of 32, 33, or 35 carbons and either zero or one degree of unsaturation. In this case the spectra show peaks corresponding to simple cationization [M+Na]+ as well as cationization accompanied with exchange of a proton with another alkali metal [M+Na2-H]+ and [M+NaK-H]+. In each case the resulting adduct ion has a net +1 charge. These ions are observed for PEs with 32:0, 33:1, and 35:1 fatty acids. Since PE contains two fatty acids, the most likely explanation is that the carbon numbers 32, 33, and 35 reflect the presence of fatty acids containing 16, 17, and 18 carbons and having 0 or 1 degree of unsaturation. While the preponderance PEs and the occurrence of a C17 fatty acid might be considered unusual in a biological system, the C16 and C17 represent about 80% of the fatty acids in E. coli. The unusual C17 fatty acid is produced biologically from a C16:1 fatty acid precursor when a cyclopropane ring forms across the double bond to give a saturated C17c having a ring rather than a double bond. This “c” notation for cyclic helps identify the location of the precursor double bond when only one is present in the phospholipid.
Figure 2. The phospholipids region of the MALDI mass spectrum (from m/z 700-800) of the organic layer obtained after a simple extraction of E. Coli.
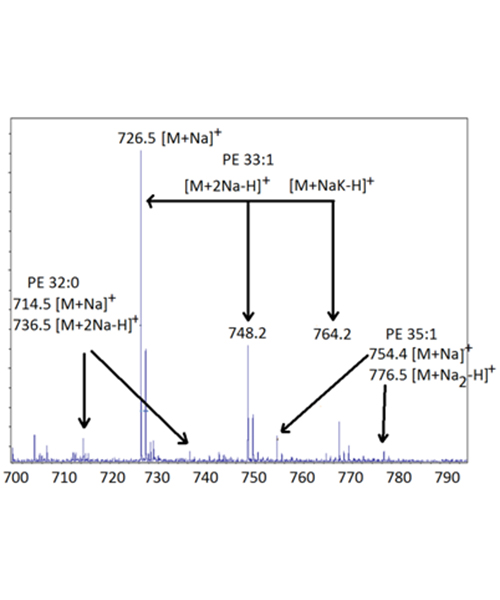
Using just the m/z values it is possible to tentatively identify the constituent fatty acids. The PE 32:0 peak consists of two C16:0 fatty acids, the PE 33:1 peak consists of a C16:0 and a C17c fatty acid, and the PE 35:1 consists of a C17c and a C18:0 fatty acid. In this case it is also possible to surmise that essentially all of the C16:1 fatty acids present in the bacterium were converted to the cyclic C17c. There is little or no signal corresponding to PE 32:1 or any other lipid containing unsaturation that is not also associated with the extra carbon to give the C17c. In other words, there are no double bonds in the fatty acids, just the novel cyclopropane rings. Identification of these peaks as PEs rather than an alternative structure can be confirmed by observation of fragment ions. These species are prone to fragmentation in the MALDI TOF MS. Either the uni-molecular or collision induced fragments can be used. In this case major fragments corresponding to loss of the neutral PE head group (and loss of C2H5N) as well as the sodium adduct ion of the PE head group confirm these are in fact PEs.
Different organisms give different characteristic and taxonomically useful mass spectra for their intact lipids (data not shown), just as would be expected based on the established utility of the FAMEs themselves for bacterial identification. However, compared to traditional GC or GC/MS and FAMEs analysis, the MALDI approach using intact lipids should be considered a rapid screening method rather than a definitive approach. The addition of the GC separation step adds time and complexity to the taxonomy measurement based on FAMEs analysis, but it also provides considerably more taxonomic power than the faster MALDI method.
Future Trends, Coupling Chromatography to MALDI MS
The utility of MALDI MS can be enhanced by addition of a chromatography step. Offline approaches include HPLC spotting of MALDI plates, HPLC with fraction collecting or direct TLC/MALDI. More recently new commercial instruments have been developed allowing direct coupling of MALDI to LC systems based on rapid drying of droplets or other novel approaches. None of these approaches has been used sufficiently for bacterial taxonomy or other applications to make definitive comparisons with the classical techniques such as FAMEs methods for bacterial taxonomy or LC/ESI MS analysis of intact polar lipids, but based on the limited data available it is clear that both approaches should improve upon the already significant utility of direct MALDI MS.
Acknowledgement: This research was support by NIH NCRR grant 5P20RR015569
References
- Gidden, J., Liyanage, R., Durham, B. and Lay, J.O. Rapid Commun. Mass Spectrom., 21, 1951-1957 (2007).
- Lay, J.O., Liyanage, R., Durham, B. and Brooks, J. Rapid Commun. Mass Spectrom., 20, 952-958 (2006).
- Gidden, J., Denson, J., Liyanage, R., Ivey, D.M. and Lay, J.O. Int. J. Mass Spectrom., 283, 178-184. (2009).
- Emmerson, B., Gidden, J., Lay, J.O. and Durham, B. J. Lipid Res., 51, 2428-2434 (2010).
- Fuchs B., Schiller J., Süss R., Zscharnack M., Bader A., Müller P., Schürenberg M., Becker M. and Suckau, D. Anal. Bioanal. Chem., 392, 849-60 (2008).
- Jones, J.J., Stump, M.J., Fleming, R.C., Lay, J.O. and Wilkins, C.L. J. Am. Soc. Mass Spectrom., 15, 1665-1674 (2004).
- Wilkins, C. L., Lay, J. O. and Winefordner, J.D. Identification of Microorgansisms by Mass Spectrometry, Series: Chemical Analysis: A Series of Monographs on Analytical Chemistry and Its Applications. (John Wiley & Sons New York (2006).
- Folch, J., Lees, M. and Stanley, G.H.S. J. Biol. Chem., 226, 479-509 (1957).
- Bligh, E.G. and Dyer, W.J. Can. J. Biochem. Physiol., 37, 911-917 (1959).